Descrição
Controle glicêmico a partir da insulinoterapia e dos hipoglicemiantes orais. Regulação dos hormônios tireoidianos e utilização dos fármacos antitireoidianos. Regulação hormonal durante o ciclo menstrual e utilização clínica dos estrógenos e progestógenos. Estimulação uterina por meio dos ocitócicos.
Propósito
Reconhecer as principais propriedades dos fármacos envolvidos com o sistema endócrino, uma vez que são frequentemente utilizados em nossa população no tratamento da diabetes, hipertireoidismo, hipotireoidismo, na reposição hormonal ou indução do parto.
OBJETIVOS
Módulo 1
Descrever as propriedades farmacológicas da insulina e dos hipoglicemiantes orais, os mecanismos de ação, as principais indicações clínicas, os efeitos colaterais e as principais contraindicações
Módulo 2
Descrever a fisiologia dos hormônios da tireoide e as propriedades farmacológicas dos fármacos antitireoidianos, os mecanismos de ação, as principais indicações clínicas, os efeitos colaterais e as contraindicações
Módulo 3
Reconhecer as propriedades farmacológicas dos estrógenos e progestógenos, os mecanismos de ação, as principais indicações clínicas, os efeitos colaterais e as contraindicações
Módulo 4
Identificar as principais propriedades farmacológicas, os mecanismos de ação, as indicações clínicas, os efeitos colaterais e as contraindicações dos ocitócicos
Introdução
O sistema neuroendócrino compreende uma complexa regulação de inúmeras funções do nosso organismo por meio de sinais transmitidos entre nossas células e nossos tecidos, sinais esses determinados por hormônios que são produzidos por diferentes órgãos e liberados na corrente sanguínea. Neste tema, abordaremos diferentes fármacos que, de alguma forma, modulam a fisiologia endócrina.
Entre os distúrbios que serão abordados, destacam-se o aumento (hiperglicemia) ou a diminuição (hipoglicemia) de glicose no organismo, que podem ocorrer por inúmeras alterações, como no funcionamento da insulina. Portanto, neste material, revisaremos a fisiologia e a farmacologia da insulina e de outros hormônios importantes que regulam a homeostasia da glicose.
Outros distúrbios frequentes encontrados na população são aqueles relacionados à tireoide. Neste tema, discutiremos brevemente a estrutura, a regulação e a fisiologia da tireoide, bem como suas principais alterações. Em seguida, abordaremos os fármacos que substituem os hormônios tireoidianos, quando estes são deficientes ou não possuem atividade adequada, bem como os fármacos que diminuem a função tiroidiana quando esta encontra-se aumentada.
Abordaremos também a fisiologia dos hormônios estrógenos e progestógenos, que desempenham inúmeros papéis fisiológicos, entre eles aqueles necessários para maturação embrionária e concepção. Atualmente, eles são muito utilizados terapeuticamente em tratamentos de reposição, para contracepção e controle dos sintomas da menopausa. Além disso, vários antagonistas são frequentemente utilizados na quimioterapia do câncer.
No final deste material, discutiremos a utilização dos ocitócicos, ou seja, fármacos que estimulam o útero. Eles são utilizados para induzir contrações uterinas semelhantes às do trabalho de parto vaginal, mas também podem ser administrados antes do parto para aumentar a dilatação do colo uterino, bem como após o parto, a fim de auxiliar na contração do útero, além de poder ser usado no tratamento da hemorragia pós-parto.
MÓDULO 1
Descrever as propriedades farmacológicas da insulina e dos hipoglicemiantes orais, os mecanismos de ação, as principais indicações clínicas, os efeitos colaterais e as principais contraindicações
Conceitos
Antes de discutirmos as propriedades farmacológicas da insulina e dos hipoglicemiantes orais, é necessário relembrarmos conceitos fisiológicos importantes para entendermos onde cada fármaco atuará.
O pâncreas é o órgão que produz insulina, hormônio responsável pela redução da glicemia, o glucagon, fator hiperglicemiante que mobiliza as reservas de glicogênio, e a somatostatina, que modula a secreção da insulina e do glucagon.
Esses hormônios são secretados pelas células das ilhotas pancreáticas (células β, α e δ, que produzem insulina, glucagon e somatostatina, respectivamente) e desempenham papéis importantes na regulação das atividades metabólicas, particularmente na homeostasia da glicose. A insulina e o glucagon constituem os dois hormônios mais importantes para o controle da homeostasia da glicose. A insulina promove o armazenamento de energia (glicose) nos tecidos-alvo, reduzindo a glicose da corrente sanguínea. De modo contrário, o glucagon atua no sentido de elevar o nível de glicemia plasmática e, consequentemente, reverter os feitos da insulina.

A insulina é uma proteína de 51 aminoácidos que consiste em duas cadeias peptídicas ligadas por pontes dissulfeto. Ela é sintetizada como um precursor (pró-insulina) que sofre hidrólise para formar a insulina e o peptídeo C. A secreção de insulina no organismo ocorre dois minutos após a ingestão de alimentos, em resposta ao aumento transitório dos níveis de glicose e aminoácidos circulantes, seguido de uma secreção pós-prandial, resposta desencadeada pelas células β.

As figuras a seguir ilustram os mecanismos de liberação de insulina, mediante o aumento da glicemia, e de captação e utilização da glicose pelas células, ações promovidas pela ligação da insulina ao seu receptor. Vale destacar que estes esquemas referem-se a indivíduos não diabéticos.
Pró-Insulina

Peptídeo C
A quantificação do peptídeo C circulante é o melhor indicador dos níveis plasmáticos de insulina, embora não desempenhe nenhuma função fisiológica conhecida.

Durante o aumento transitório de glicose, há elevação dos níveis intracelulares de ATP, que fecham os canais de potássio dependentes de ATP. A diminuição do efluxo de potássio resulta na despolarização da célula β e na abertura dos canais de cálcio regulados por voltagem, o que desencadeia a secreção do hormônio, mecanismo que é explorado por vários fármacos, como as sulfonilureias e as meglitinidas.

Após a liberação da insulina, ela se liga ao seu receptor e estimula a captação e a utilização da glicose, por exemplo, nas células do fígado, tecido adiposo e músculo esquelético. No entanto, essa regulação fisiológica é alterada em pacientes diabéticos.
Assim, podemos dizer que o paciente com diabetes é aquele que apresenta níveis elevados de glicemia associados a uma secreção de insulina inadequada ou ausente, ou com comprometimento de sua ação.
Atualmente, existem quatro classificações clínicas do diabetes. Vamos conhecer cada uma delas a seguir.
O DM1, também denominado diabetes melito insulinodependente, manifesta-se mais em crianças, adolescentes e adultos jovens, sendo caracterizado por deficiência absoluta de insulina devido à destruição autoimune das células β. Nesses casos, o pâncreas deixa de responder à glicose, e o paciente apresenta sintomas clássicos, como polidipsia, polifagia, poliúria e perda de massa corporal. O paciente com DM1 não consegue manter um nível de secreção basal de insulina nem responder às variações de glicose circulantes (Figura 3). Para os pacientes com DM1, a terapia de reposição com insulina é necessária para a manutenção da vida.
O DM2, também denominado diabetes melito não insulinodependente, corresponde a mais de 90% dos casos e é influenciado por fatores genéticos, como idade, obesidade e resistência à insulina nos tecidos periféricos. O paciente com DM2 apresenta uma secreção de insulina insuficiente pelo pâncreas (Figura 3), além de diminuição da quantidade de células β ao longo do tempo. A indicação para restrição calórica e atividades físicas está associada ao aumento na responsividade à insulina. Portanto, somente quando as alterações no estilo de vida não são suficientes para a correção da concentração de glicose, é indicado o uso dos hipoglicemiantes orais.
Fisiologicamente, durante a gravidez, a gestante desenvolve resistência à insulina. O diabetes gestacional é definido como qualquer anormalidade dos níveis de glicose observada pela primeira vez durante a gravidez, e, se não for controlada, pode causar má formações fetais.
São exemplos desse tipo de diabetes a pancreatite e as reações após terapia farmacológica.
Conforme discutimos anteriormente, as alterações no funcionamento da insulina podem causar grave hiperglicemia. Se não for tratada, pode resultar em retinopatia, nefropatia, neuropatia e complicações cardiovasculares. Assim, a administração de insulina exógena ou outro fármaco hipoglicemiante é extremamente importante, uma vez que pode reduzir a morbidade e a mortalidade associadas ao diabetes.

Preparações de insulina exógena
O principal objetivo da utilização da insulina exógena é substituir a ausência de secreção da insulina endógena no paciente com DM1 ou para suplementar a secreção insuficiente de insulina endógena no paciente com DM2, quando a dieta e outras formas de terapia são insuficientes para controlar a hiperglicemia. A meta da insulinoterapia consiste em reproduzir a secreção fisiológica de insulina no indivíduo saudável e repor a insulina basal (noturna, em jejum e entre as refeições), bem como a insulina em bolo ou prandial (durante as refeições).
A insulina exógena é atualmente produzida por biotecnologia (técnica de DNA recombinante). Modificações em sua estrutura foram capazes de produzir insulinas com propriedades farmacocinéticas distintas, mas preservando a especificidade pelo seu receptor. Por ser um polipeptídeo, a insulina exógena é rapidamente degradada no trato gastrintestinal (TGI), sendo imprópria para administração via oral – logo, é frequentemente realizada por injeções subcutâneas (SC). A infusão SC contínua de insulina (bomba de insulina) é uma alternativa que pode ser mais conveniente para alguns pacientes, eliminando as múltiplas injeções diárias de insulina.
As preparações de insulina são classificadas de acordo com sua duração de ação, em preparações de ação curta e de ação longa. O gráfico a seguir ilustra o início e a duração de ação das insulinas descritas anteriormente.

Além das modificações na estrutura da insulina, que resultam em alterações na absorção e nos efeito dos fármacos, variações na forma de apresentação também modificam as propriedades farmacocinéticas.
Exemplo
A insulina preparada em forma de solução (regular) já está completamente dissolvida e é rapidamente absorvida. Já as insulinas preparadas em suspensão dissolvem-se lentamente no tecido subcutâneo e promovem um efeito mais prolongado. É importante salientarmos que dose, local de injeção, fluxo de sangue, temperatura e atividade física também podem afetar o início e a duração de ação da insulina.
Baseado nestes conceitos, começaremos agora a estudar as principais propriedades farmacológicas das preparações de insulina.
Conheça, a seguir, as insulinas de ação curta. São elas:
São classificadas como insulinas de ação muito rápida, com o início de ação entre 30 e 90 minutos e duração efetiva de três a quatro horas.
Também denominada insulina em bolo prandial clássica, possui seu início de ação entre 120 e 180 minutos e apresenta duração efetiva de quatro a seis horas.
De forma geral, as insulinas de ação curta são administradas para mimetizar a liberação de insulina durante as refeições e controlar a glicose pós-prandial. Essas insulinas são frequentemente usadas em associação com uma insulina basal de ação mais longa, responsável por controlar a glicemia de jejum. Elas também (preferencialmente a insulina regular) podem ser utilizadas em emergências, para casos em que são necessárias correções rápidas de glicose elevada, como no coma hiperglicêmico.
Orienta-se administrar a insulina regular por via subcutânea (SC) 30 minutos antes das refeições, ao passo que as outras insulinas de ação curta podem ser administradas 15 minutos antes.
Conheça, a seguir, as insulinas de ação longa. São elas:
A insulina isófana, ou insulina neutra com protamina hagedorn (NPH), foi desenvolvida pela adição de zinco e protamina à insulina regular, o que culminou em um complexo menos solúvel, resultando em retardo na absorção. Ela apresenta um início de ação tardio em cerca de duas a seis horas com duração de 10 a 16 horas. É usada para controle basal (jejum) nos DM1 e DM2 e, em geral, é administrada com a insulina de ação curta durante a refeição. Como existe intensa variabilidade de absorção, combinada com a disponibilidade de outros análogos de ação longa com ação mais previsível, seu uso clínico tem diminuído.
A insulina glargina promove a formação de um precipitado no local da injeção, o qual libera insulina por um período prolongado. Tem início mais lento (uma a quatro horas) do que a insulina NPH e um efeito hipoglicêmico achatado e prolongado, sem pico, que pode variar de 12 a 24 horas (Figura 3).
A insulina detemir é a que apresenta o efeito mais reproduzível entre as insulinas de ação longa, com início de ação de uma a quatro horas, com duração de ação entre 12 e 20 horas.
Outra insulina de ação longa é a degludeca, que apresenta o início de ação entre uma e quatro horas, com duração efetiva de 24 a 42 horas, e provoca hipoglicemia menos grave em comparação à insulina glargina.
Os esquemas terapêuticos da utilização da insulina exógena – incluindo preparação, dose e frequência de administração – devem ser individualizados para cada paciente e, com frequência, são ajustados ligeiramente a cada dia, de acordo com a atividade do paciente, quantidade e composição das refeições e níveis de glicemia.

Caso não responda ao tratamento padrão (controle glicêmico) com duas, três, ou até mais injeções diárias de insulina de ação longa, com o intuito de diminuir o número de injeções diárias e melhorar o controle de glicose do paciente, o médico pode realizar associações entre as insulinas de curta ação e de ação longa descritas. No mercado brasileiro, existem disponíveis diversas preparações, como 70% da insulina NPH associada a 30% insulina regular.
Dentre os efeitos adversos do uso da insulina exógena, a hipoglicemia é a mais comum e grave, entretanto existem outros efeitos relatados, como aumento de massa corporal, reações alérgicas no local da injeção e lipodistrofia, que pode ser minimizada por rotação do local de aplicação. Os diabéticos com insuficiência renal podem precisar de redução da dose de insulina.
Conforme já abordado, o principal objetivo da insulinoterapia é mimetizar a secreção da insulina secretada no indivíduo saudável. Para isso, muitas vezes, a administração de uma insulina de longa ação à noite gera um nível basal, enquanto uma insulina de efeito rápido é utilizada antes das refeições, sendo necessária associação das insulinas.
Hipoglicemia
Paciente apresenta-se taquicárdico, com agitação, tremor, palidez e sudorese intensa.
Lipodistrofia
Atrofia ou hipertrofia do tecido gorduroso subcutâneo, localizado no local da injeção.
Hipoglicemiantes orais
Esses fármacos são úteis no tratamento do DM2 não controlado com dieta e atividade física. Os pacientes que são diagnosticados com diabetes com menos de cinco anos ou após os 40 anos de idade respondem melhor ao uso destes fármacos. Não há nenhum mecanismo de ação comum a todos os hipoglicemiantes orais, por isso eles serão abordados baseados em seu mecanismo de ação específico.
A metformina, medicamento da classe das biguanidas, é o fármaco de escolha para o tratamento de pacientes com DM2, e é classificado como um sensibilizador à insulina. Aproximadamente 70% dos pacientes fazem uso de metformina como monoterapia ou em associação a outro fármaco, principalmente em indivíduos obesos. Seu mecanismo de ação não é totalmente compreendido, mas já foi observada redução da gliconeogênese hepática (inibição da produção hepática de glicose – efeito principal), redução da absorção intestinal de açúcar e aumento da captação e uso de glicose pelos tecidos-alvo, diminuindo a resistência à insulina. Ela não aumenta a liberação de insulina e, por isso, não promove hiperinsulinemia. É bem absorvida por via oral, não se liga a proteínas plasmáticas e é eliminada inalterada pela urina.

Metformina
Os principais efeitos adversos da metformina estão relacionados ao TGI, como anorexia, náuseas, desconforto abdominal e diarreia, no entanto não há risco de hipoglicemia. Ela é contraindicada para pacientes idosos e aqueles que apresentam insuficiência renal, devido ao risco de acidose láctica, e deve ser suspensa em casos de infarto agudo do miocárdio, insuficiência cardíaca ou sepse.
Esses fármacos estimulam a secreção de insulina das células β do pâncreas, mimetizando o mecanismo de liberação endógeno. A ligação destes fármacos, derivados da sulfonilureia, ao seu receptor inibe o efluxo de íons de potássio, com consequente despolarização da membrana. Essa despolarização abre um canal de cálcio regulado por voltagem, resultando em influxo de cálcio e liberação da insulina pré-formada. Além disso, eles também podem diminuir a produção de glicose pelo fígado e aumentar a sensibilidade periférica à insulina. Em geral, as sulfonilureias são eficazes, seguras e baratas e, associadas a metformina, constituem o principal tratamento do DM2.
Administradas por via oral, ligam-se às proteínas plasmáticas, e, assim, podem apresentar interações medicamentosas com outros fármacos que também se ligam fortemente às proteínas, como as sulfonamidas e o AAS. São biotransformados pelo fígado e excretadas pelo fígado e pelos rins. A duração de ação varia de 12 a 24 horas. A glipizida é a que possui meia-vida mais curta (duas a quatro horas), e deve ser ingerida 30 minutos antes do desjejum, visto que sua absorção é retardada quando ingerida com alimentos.
Os principais efeitos adversos são hipoglicemia, aumento de massa corporal e hiperinsulinemia, dada a secreção excessiva de insulina. A glibenclamida é contraindicada na presença de comprometimento hepático, bem como em pacientes com insuficiência renal. Por atravessar pouco a placenta, porém, pode ser uma alternativa à insulina em gestantes.

Glibenclamida

Glipizida

Glimepirida
Esses fármacos, da classe meglitinida, apresentam o mesmo mecanismo de ação das sulfonilureias, promovendo a liberação de insulina das células β do pâncreas, mas diferem na estrutura química, e assim apresentam início de ação mais rápida e duração mais curta. Elas são particularmente eficazes na liberação precoce de insulina que ocorre depois da refeição e, desse modo, podem ser ingeridas logo antes da refeição.
Dessa forma, estão indicadas para uso no controle da glicose pós-prandial.
Devem ser administradas imediatamente antes das refeições e são bem absorvidas após administração oral. São biotransformadas em produtos inativos pelo CYP3A4 no fígado e são excretadas pela bile.
Atenção
Atenção à utilização de fármacos que possam inibir a enzima CYP3A4 (Ex.: antirretrovirais, antifúngicos imidazólicos e a pioglitazona) e aumentar o feito hipoglicemiante, ou induzir a enzima (Ex.: barbitúricos e carbamazepina) e reduzir o efeito hipoglicemiante.
São contraindicadas em associação às sulfonilureias, devido à sobreposição dos mecanismos de ação, e à genfibrozila (fármaco anti-hiperlipemiante), por aumentar seu efeito. Embora com pouca incidência, os principais efeitos adversos são hipoglicemia e aumento de massa corporal. Esses fármacos podem ser usados em pacientes com insuficiência renal e em idosos.

Repaglidina

Nateglinida
As tiazolidinadionas alteram a expressão de vários genes envolvidos no metabolismo dos lipídeos e da glicose, na transdução de sinais de insulina e na diferenciação dos adipócitos e de outros tecidos, resultando em aumento da sensibilidade da ação da insulina no tecido adiposo, no fígado e no músculo esquelético e aumentam a captação celular de glicose. A pioglitazona é recomendada como um fármaco de segunda ou terceira escolhas para o DM2. A rosiglitazona tem sido descontinuada devido a inúmeros efeitos adversos cardíacos.
Além do efeito no controle da glicose, a rosiglitazona aumenta a lipoproteína de baixa densidade (LDL-C) e os triglicerídeos, ao passo que a pioglitazona diminui os triglicerídeos. Os dois fármacos aumentam a lipoproteína de alta densidade (HDL-C).
São bem absorvidas após administração por via oral e são extensamente ligadas à albumina plasmática. Ambas sofrem extensa biotransformação por diferentes isoenzimas do sistema CYP450 e são consideradas indutores enzimáticos. Os efeitos adversos incluem retenção de líquidos, insuficiência cardíaca, toxicidade hepática, aumento de massa corpórea e fraturas ósseas. Esses fármacos são contraindicados em lactantes e em pacientes com insuficiência cardíaca grave.
Atenção
Atenção aos fármacos que são metabolizados pela enzima CYP450, como, por exemplo, os contraceptivos orais com estrogênio, que podem não apresentar efeito farmacológico devido à indução de sua metabolização; assim, é aconselhável a utilização de outros métodos de contracepção durante a terapia com pioglitazona.

Pioglitazona

Rosiglitazona
A acarbose é indicada para o tratamento do DM2 e mostra-se efetiva quando administrada com as refeições, mas não em outros horários. Ela inibe reversivelmente a enzima α-glicosidase (localizada na borda do epitélio intestinal), que libera glicose dos dissacarídeos, aumentando a absorção. Portanto, esse fármaco inibe a absorção da glicose, reduzindo os níveis de glicose pós-prandial. Ela precisa ser biotransformada pelas bactérias intestinais para tornar-se ativa, e é excretado pela urina.
É importante salientar que, quando o paciente apresenta hipoglicemia durante o tratamento com a acabose, é necessário que ela seja tratada com a administração de glicose, pois a hidrólise da sacarase (frequentemente utilizada) também é inibida por esses fármacos.
Os principais efeitos adversos são flatulência, distensão, desconforto abdominal, diarreia e cólicas intestinais, efeitos que, muitas vezes, não são tolerados pelos pacientes, tornando a utilização limitada na prática clínica. Tais efeitos resultam em aparecimento de carboidratos não digeridos no cólon, que são então fermentados em ácidos graxos de cadeia curta, com liberação de gases. A acarbose é contraindicada em pacientes com alterações gastrointestinais e insuficiência renal.

Acarbose
Esses fármacos inibem a enzima dipeptidilpeptidase-4 (DPP-4), que é responsável pela inativação dos hormônios incretina (GLP-1, descrito anteriormente). Essa inibição culmina em aumento da liberação de insulina em resposta às refeições. Possuem indicação clínica para o tratamento do DM2, são bem absorvidos após administração por via oral e são excretadas na urina. Assim, exigem o ajuste de dose em pacientes com insuficiência renal. Os efeitos adversos mais comuns são nasofaringite e cefaleia, mas, em geral, são fármacos bem tolerados.

Alogliptina

Linagliptina

Saxagliptina

Sitagliptina
São fármacos utilizados no tratamento do DM2, e o mecanismo de ação envolvido é a inibição do cotransportador sódio-glicose 2 (SGLT2), responsável por 90% da reabsorção de glicose filtrada nos rins. Dessa forma, diminuem a reabsorção renal de glicose, aumentam a excreção urinária e diminuem a glicemia. Também diminuem a reabsorção de sódio e promovem diurese osmótica. Desse modo, podem reduzir a pressão arterial, embora não sejam indicados no tratamento da hipertensão. Esses fármacos são administrados uma vez ao dia, pela manhã, antes da primeira refeição. São contraindicados em pacientes com insuficiência renal. Os efeitos adversos mais comuns são a candidíase vulvovaginal e infecções do trato urinário.

Canagliflozina

Dapagliflozina
Saiba mais
Outros fármacos, como a pranlintida, a bromocriptina e colesevelam, podem ser utilizados como hipoglicemiantes orais, mas atuam por outros mecanismos ou mecanismos pouco definidos. Por apresentarem pouca eficácia na redução dos níveis de glicose, seu uso é questionável.
O vídeo aborda as diferenças nas aplicações da insulina e dos hipoglicemiantes orais.
Miméticos da incretina: exenatida, lixisenatida e liraglutida
Fisiologicamente, os hormônios incretina são liberados no intestino após uma carga de glicose oral e são responsáveis por 60 a 70% da secreção pós-prandial de insulina. Dentre eles, destaca-se o hormônio peptídeo, semelhante a glucagon 1 (GLP-1). A exenatida, lixisenatida e a liraglutida mimetizam o efeito deste hormônio (fármacos incretinomiméticos – agonistas do GLP-1) e são usados no tratamento do DM.

Exenatida

Lixisenatida

Liraglutida
Esses fármacos melhoram a secreção de insulina dependente de glicose, retardam o esvaziamento gástrico, reduzem o apetite, diminuem a secreção pós-prandial de glucagon e promovem a proliferação de células β. Diferentemente do que ocorre com a insulina, os pacientes apresentam perda de peso de dois a três quilos com a utilização desses fármacos, o que contribui para a melhoria do controle da glicose. Por isso, são fármacos de escolha para o tratamento de DM2 em pacientes obesos e com síndrome metabólica.
Esses fármacos também precisam ser administrados por via SC. A liraglutida apresenta meia-vida longa, permitindo apenas uma dose diária. Já a exenatida apresentada em canetas de doses fixas possui meia-vida mais curta e precisa ser injetada duas vezes ao dia dentro de 60 minutos antes do desjejum e do jantar, e deve ser evitada por pacientes com insuficiência renal grave. Os principais efeitos adversos são náuseas, êmeses, diarreia e constipação, e podem aumentar o risco de pancreatite.
Entre todos os fármacos abordados neste módulo, alguns pacientes vão utilizá-los em monoterapia. Entretanto, outros podem exigir uma combinação de hipoglicemiantes orais, com ou sem insulina, para controlar a hiperglicemia, efeito que é garantido pela associação de distintos alvos moleculares e mecanismos de ação. Assim, devemos estar atentos aos efeitos adversos, como hipoglicemia, pois, em muitos casos, é possível usar menor dose de cada fármaco, reduzindo, assim, os efeitos adversos.
Saiba mais
Alguns fármacos abordados anteriormente de forma interessante, como a metformina e as tiazolidinadionas, além de serem utilizados no controle da glicemia, também são eficazes no tratamento da síndrome do ovário policístico. Eles diminuem a resistência à insulina observada nesse distúrbio e podem normalizar a ovulação.
Atualmente, pesquisas têm sido realizadas com o intuito de desenvolver terapias-alvo para reverter a disfunção das células β, para o tratamento dos pacientes com DM1. Além disso, estão sendo desenvolvidos agentes que inibem enzimas da síntese de glicogênio e glicogenólise para limitar a produção de glicose ou até mesmo facilitar a excreção da glicose no túbulo proximal renal, com foco no tratamento para a DM2.
Verificando o aprendizado
ATENÇÃO!
Para desbloquear o próximo módulo, é necessário que você responda corretamente a uma das seguintes questões:
O conteúdo ainda não acabou.
Clique aqui e retorne para saber como desbloquear.
MÓDULO 2
Descrever a fisiologia dos hormônios da tireoide e as propriedades farmacológicas dos fármacos antitireoidianos, os mecanismos de ação, as principais indicações clínicas, os efeitos colaterais e as contraindicações
Conceitos
Neste módulo, reconheceremos as vias e os mecanismos de regulação dos hormônios sintetizados na tireoide, bem como as ações destes. Abordaremos também as principais características farmacológicas da terapia utilizada para os distúrbios mais frequentes da tireoide, como o hipotireoidismo e o hipertireoidismo, também denominado de tireotoxicose.
Tri-iodotironina e tetraiodotironina

Tri-iodotironina (T3)

Tetraiodotironina (T4)

A glândula tireoide é responsável pela produção de hormônios essenciais, que exercem efeitos variados sobre a homeostasia metabólica. Entre os hormônios produzidos pelas células foliculares da tireoide, destacam-se a tri-iodotironina ou liotironina (T3, a forma mais ativa) e a tiroxina ou tetraiodotironina (T4) (Figura 7), que possuem, em sua estrutura, muitas moléculas de iodo associadas.
Tri-iodotironina (T3)
Tetraiodotironina (T4)
Quando liberados no organismo, os hormônios tireoidianos possuem inúmeras ações fisiológicas em diferentes órgãos.

A produção inadequada desses hormônios – condição denominada hipotiroidismo, e, em casos graves, mixedema –está associada a inúmeros sintomas, entre eles bradicardia, fala arrastada, letargia, baixa resistência ao frio, retardo mental, físico e nanismo, principalmente em crianças. A deficiência congênita desse hormônio resulta em cretinismo, forma de retardo mental grave, porém passível de prevenção. Além disso, os pacientes também podem desenvolver um espessamento característico da pele. Em contraste, o excesso de síntese e secreção desses hormônios (hipertireoidismo) está associado a sintomas como:
- Taquicardia.
- Arritmias.
- Nervosismo.
- Tremores.
- Produção excessiva de calor.
- Sudorese.
Letargia
Um estado de cansaço que envolve diminuição da energia, da capacidade mental e da motivação.
Precisamos entender que a produção desses hormônios é altamente regulada. A síntese de hormônios pela tireoide ocorre a partir da liberação do hormônio que disponibiliza tireotrofina (TRH), que é liberado pelo hipotálamo e estimula a hipófise a sintetizar e liberar o hormônio estimulante da tireoide (TSH), que, por sua vez, estimula a tiroide a produzir T3, T4 e calcitonina. A partir de um mecanismo de retroalimentação negativa, a secreção de TRH e TSH diminui à medida que os níveis plasmáticos de T3 e T4 aumentam, ou seja, a produção dos hormônios é ajustada “de forma automática” à demanda, conforme ilustrado na figura a seguir.

O fluxo apresenta as principais etapas de síntese:
Armazenamento e secreção dos hormônios tireoidianos ocorrem a partir da liberação do TSH, que induz à captação de iodeto (I-) plasmático pelas células foliculares da glândula tireoide. Em seguida, sofre oxidação a iodo (I2) e é incorporado a tiroglobulina, processo denominado organificação do iodeto.
As reações são catalisadas pela tireoide-peroxidase. Há, então, acúmulo, ou seja, a formação de um grande estoque de tireoglobulina iodada dentro da glândula.
Após diferentes formas de condensação dessas moléculas e a posterior clivagem proteolítica da tireoglobulina, há formação e secreção dos hormônios T3 e T4.
Esses hormônios são constituídos por uma estrutura de duas moléculas de tirosina iodadas, unidas por uma ligação éter.
Esses hormônios são frequentemente encontrados no plasma, ligados a proteínas plasmáticas, principalmente a globulinas, e precisam estar em sua forma livre antes de entrar nas células e promover sua atividade metabólica. Neste sentido, diversos fármacos e alguns distúrbios podem alterar a ligação desses hormônios tireoidianos às proteínas plasmáticas, modificando sua eficácia. Quando se encontra em sua forma livre e no interior das células, o hormônio T4 (pró-hormônio) é convertido em T3, e é capaz de entrar no núcleo, ligar-se a regiões específicas no DNA e alterar a expressão gênica das células, produzindo proteínas específicas, responsáveis pelos efeitos dos hormônios tireoidianos.

Conforme já abordamos, a estrutura química desses hormônios é composta por inúmeras moléculas de iodo. Assim, o consumo adequado de iodo é essencial para a produção normal dos hormônios tireoidianos.

No entanto, quando o aporte de iodo no organismo é baixo, há redução na síntese dos hormônios tireoidianos e, a partir de um mecanismo de regulação, há aumento excessivo da secreção de TSH. Isso estimula a tireoide a trabalhar em baixas concentrações de iodo, tornando-a hiperplásica e hipertrófica. Consequentemente, há aumento de seu volume e tamanho (papeira ou também denominado bócio).

Legumes, carne vermelha e aves contêm quantidades mínimas de iodo, enquanto laticínios e peixes apresentam um teor de iodo relativamente alto.
Neste contexto, em algumas regiões, o solo é deficiente em iodo e, consequentemente, o consumo também é reduzido. Nessas regiões, é frequente o aumento dos casos de bócio, caracterizado por deficiência alimentar de iodo. No sentido de evitar esse quadro, atualmente existe a profilaxia com o sal de cozinha iodado, no intuito de suprir as necessidades de iodo e evitar, de forma simples e eficaz, a ocorrência de bócio.
Baseado nestes conceitos, começaremos agora a estudar as principais propriedades farmacológicas e as indicações clínicas dos hormônios tireoidianos, muitas vezes utilizados em reposição hormonal, e dos medicamentos antitireoidianos.
Liotironina (T3) e Levotiroxina (T4)
Quando é necessária a reposição do hormônio tireoidiano deficiente, como em pacientes com hipotireoidismo e com hipotireoidismo autoimune –também denominado tireoidite de Hashimoto –o tratamento pode ser feito com administração oral de hormônios da tireoide. Os fármacos são produzidos por síntese química e estruturalmente idênticos aos hormônios tireoidianos endógenos. Entre esses fármacos, tanto a liotironina sódica (T3) quanto a levotiroxina sódica (T4) são bem absorvidos por via oral. O fígado constitui o principal local de degradação dos hormônios e, antes de serem excretados na bile e em parte na urina, os hormônios sofrem conjugação com glicuronídeos e sulfatos.

As principais indicações clínicas desses hormônios sintéticos estão voltadas para o tratamento do hipotiroidismo, que geralmente é ocasionado pela destruição autoimune da glândula ou ausência da função da enzima peroxidase, responsável pela síntese dos hormônios.
Embora o T3 seja a forma ativa e mais bem absorvida pelo intestino, a utilização da levotiroxina sódica (T4) é preferida quando comparada à liotironina sódica (T3) ou até mesmo a fármacos produzidos com a associação dos hormônios T3 e T4, denominado liotrix.
A utilização da levotiroxina sódica (T4) é preferível, pois apresenta maior meia-vida, maior estabilidade, obtendo concentrações plasmática mais estáveis, pois a degradação é mais lenta. Assim, o fármaco pode ser administrado uma vez ao dia, e o equilíbrio é alcançado entre seis e oito semanas. Além disso, a determinação laboratorial durante o uso da levotiroxina é mais fácil, e estudos mostram maior benefício em ter um grande reservatório de “pro-fármaco” tireoidiano (T4) no plasma. Em certas ocasiões, a liotironina (T3) pode ser utilizada quando se deseja um início de ação mais rápido.
As concentrações plasmáticas de TSH e de tiroxina livre devem ser sempre determinadas antes da administração da levotiroxina sódica (T4) para evitar alterações transitórias nos níveis séricos.
Embora sejam bem absorvidos por via oral, precisamos ficar atentos à interação desses fármacos com alimentos (Ex.: farelo, soja, café), com a flora intestinal, com preparações com cálcio e antiácidos contendo alumínio, pois eles diminuem a absorção da levotiroxina sódica (T4). Além disso, outros fármacos que induzem enzimas do CYP450, como rifampicina, fenobarbital, carbamazepina, fenitoína e antirretrovirais, podem acelerar o metabolismo dos hormônios da tireoide e diminuir sua eficácia.
Considerando as interações anteriores, orienta-se que o paciente administre a levotiroxina sódica (T4) 60 minutos antes do café da manhã, quatro horas depois das refeições ou ao se deitar, pois sua absorção é melhor no estômago vazio. A acidez gástrica fisiológica também é necessária para a correta absorção da levotiroxina sódica (T4), e, neste sentido, orienta-se aumentar a dose do fármaco em pacientes infectados por Helicobacter pylori ou que utilizam os inibidores da bomba de prótons (Ex.: omeprazol).
A levotiroxina está disponível em comprimidos e em cápsulas preenchidas com líquido para administração oral e em forma de pó liofilizado para injeção. Estudos observaram que a apresentação da levotiroxina em cápsula de gel macia apresentou dissolução mais rápida e mais completa, além de ter sido menos afetada pelo pH gástrico ou pelo café quando comparada à formulação em comprimidos. Atualmente, a forma farmacêutica mais utilizada é a de comprimido simples.
O início do tratamento da reposição dos hormônios tireoidianos, deve ser realizado com baixas doses da levotiroxina sódica (T4), seguido de um aumento gradual para evitar indução muito rápida do metabolismo e, consequentemente, sobrecarregar o sistema cardiovascular.
Os efeitos adversos mais comuns que os pacientes apresentam durante a reposição com os hormônios tireoidianos são nervosismo, palpitações, taquicardia, intolerância ao calor e perda inexplicada de massa corporal, associados a produção fisiológica aumentada destes hormônios. Esses efeitos assemelham-se aos sintomas apresentados por pacientes com hipertireoidismo. A eficácia do tratamento pode ser monitorada a cada seis meses ou um ano a partir de dosagens dos níveis plasmáticos de TSH e hormônio tireoidiano.
Conforme já abordado anteriormente, diversos pacientes apresentam aumento na produção dos hormônios da tireoide, condição denominada como hipertiroidismo, ou doença de Graves (doença autoimune), podendo ocasionar até crises tireotóxicas. Nesses pacientes, há produção de um autoanticorpo IgG específico que se liga ao receptor de TSH, conhecido como imunoglobulina estimulante da tireoide. Esse anticorpo atua como agonista, ativando o receptor de TSH e, portanto, estimulando a síntese e a liberação de hormônio tireoidiano pelas células foliculares da tireoide.
O tratamento desses pacientes tem como principal objetivo a redução da síntese e/ou redução da liberação excessiva desses hormônios, que pode ser induzida por terapia farmacológica com os fármacos antitireoidianos, ou por meio da remoção parcial ou total da glândula tireoide, seja via cirurgia, seja por administração de iodo-131 (131I). O 131I é administrado por via oral, captado seletivamente pela tireoide e induz ao dano seletivo das células; emite radiação β de curto alcance e, dessa maneira, afeta apenas as células foliculares da tireoide. Em seguida, os pacientes apresentam o quadro de hipotireoidismo e necessitam realizar terapia de reposição com os hormônios tireoidianos descritos anteriormente.
Abordaremos, agora, as propriedades dos principais fármacos antitireoidianos.

Propiltiouracila

Metimazol
Os fármacos antitireoidianos, derivados das tioaminas, não possuem um mecanismo de ação completamente compreendido, mas são caracterizados por inibir a tireoide-peroxidase, reduzindo a incorporação do iodo à tiroglobulina, bloqueando, desse modo, a síntese e a liberação dos hormônios da tireoide. Portanto, esses fármacos são antagonistas de T3 e T4, mas, por não induzirem efeitos na tiroglobulina já armazenada na glândula, seus efeitos clínicos podem demorar de três a quatro semanas para ocorrer, até que as reservas de tiroglobulina estejam esgotadas.
O grupo tiocarbamida (S-C-N) é essencial para a atividade antitireoidiana. Em geral, a utilização desses fármacos tem sido eficaz no controle do hipertireoidismo, e muitos pacientes sofrem remissão no período de seis meses a um ano após o início do tratamento, conseguindo manter os níveis fisiológicos dos hormônios tireoidianos após a suspensão dos medicamentos.
Esses fármacos são bem absorvidos após administração oral, porém em taxas variáveis, acumulando-se rapidamente na tireoide. Após serem metabolizados, são eliminados, preferencialmente na urina.
O metimazol é o fármaco preferido quando comparado a propiltiouracila, porque é cerca de dez vezes mais potente, apresentando meia-vida mais longa e permitindo uma dosificação ao dia, apresentando menor incidência de efeitos adversos. Além da propiltiouracila apresentar meia-vida curta (duas a três horas em dosagens normais), que exige a sua administração três vezes ao dia, e, por isso, dificultar a adesão ao tratamento, sua utilização está associada a maior incidência de efeitos adversos.
Atualmente, o tiamazol também tem sido muito utilizado, seguindo as mesmas propriedades farmacológicas do metimazol. Além destes, em alguns países, há liberação do carbimazol, um pró-farmaco que é convertido em metimazol in vivo.

As tioaminas são geralmente bem toleradas, e o efeito adverso mais comum é o exantema pruriginoso (erupções comuns, restrito somente ao início do tratamento). A propiltiouracila foi associada a hepatotoxicidade e a neutropenia, agranulocitose e vasculite – reações raras, porém graves. Dessa forma, orienta-se sempre realizar a monitorização da leucometria desses pacientes e orientá-los a avisar, caso apresentem algum sintoma, principalmente dor de garganta. Com frequência, o tratamento com as tioaminas resulta em artralgias e formação de bócio, sendo, portanto, comumente designados como bociógenos.
O metimazol é contraindicado durante o primeiro trimestre da gestação, devido ao risco maior de efeitos teratogênicos (ocorrência de embriopatia), sendo preferível a utilização da propiltiouracila. Em seguida, o metimazol é usado durante o restante da gestação devido à preocupação de insuficiência hepática associada à propiltiouracila. Ambos os fármacos são secretados em baixas concentrações no leite materno, porém são considerados seguros durante o aleitamento.
Esse fármaco é um inibidor da captação de iodeto até a célula folicular da tireoide, o que resulta em redução da quantidade de iodeto disponível para a síntese dos hormônios tireoidianos. Seus efeitos não são imediatos, devido à reserva de hormônios já formados na glândula. Atualmente, seu uso é raro para fins clínicos, em razão da associação com o desenvolvimento de anemia aplásica.
Antes da introdução clínica das tioamidas, o iodeto inorgânico era o principal agente antitireoidiano; no entanto, sua utilização, hoje, como monoterapia é rara.
O iodeto inorgânico estável em dosagem elevada exerce efeito antitireoidiano temporário no hipertireoidismo, e o efeito estabelece-se mais rapidamente do que com os outros fármacos. As principais indicações clínicas incluem a supressão pré-cirúrgica da secreção da tireoide, visto que ele diminui o tamanho e a vascularidade da glândula tireoide, além da consistência do órgão ficar mais firme. Em geral, o iodo é administrado por via oral em soluções de iodeto de potássio (“iodo de Lugol”).
De forma interessante, existem alguns relatos na literatura da utilização do iodeto inorgânico como prevenção. Por exemplo, após o acidente nuclear de Chernobyl, com o intuito de evitar a captação do iodeto radioativo ambiental, seguido de destruição da tireoide em nível populacional, milhares de crianças iniciaram a administração de altas doses de iodeto durante alguns dias para suprimir temporariamente a função da tireoide, ou seja, a absorção do iodo radioativo, evitando danos à fisiologia do órgão.
Os principais efeitos adversos da administração de iodo são reações alérgicas, como angioedema, erupções cutâneas e febre medicamentosa. Sua utilização é contraindicada em terapia de longo prazo para hipertireoidismo e em tireotoxicose induzida por iodo. Além disso, sua utilização é contraindicada durante a gravidez, visto que ele atravessa a placenta e pode causar bócio no feto.
Também existem relatos da utilização dos íons lítio no lugar do iodo para a supressão rápida da tireoide no hipertireoidismo induzida por iodo, pois inibem a via de liberação de tireoxina. Outras evidências apontam que o lítio também pode inibir a síntese desse hormônio. Entretanto, o mecanismo responsável por tais ações não é conhecido.
Assista, no vídeo, às condições clínicas em que os hormônios tireoidianos são empregados e o uso deles off-label para emagrecimento.
Verificando o aprendizado
ATENÇÃO!
Para desbloquear o próximo módulo, é necessário que você responda corretamente a uma das seguintes questões:
O conteúdo ainda não acabou.
Clique aqui e retorne para saber como desbloquear.
MÓDULO 3
Reconhecer as propriedades farmacológicas dos estrógenos e progestógenos, os mecanismos de ação, as principais indicações clínicas, os efeitos colaterais e as contraindicações
Conceitos
Inicialmente, precisamos relembrar como ocorre a produção fisiológica dos estrógenos e progestógenos. Em seguida, discutiremos os principais fármacos que mimetizam as funções dos hormônios endógenos ou atuam em seus receptores. Além disso, compreender o ciclo ovariano ou menstrual proporciona uma base para o entendimento da farmacologia da contracepção, que será abordada neste módulo.
Atenção
Não podemos esquecer que o sistema endócrino é regulado pelo SNC e que as células presentes no hipotálamo sintetizam e liberam moléculas que induzem a liberação de hormônios pela hipófise, que são liberados na corrente sanguínea e distribuídos ao organismo. Neste sentido, o hormônio liberador de gonadotrofina (GnRH), secretado pelo hipotálamo em pulsos, estimula a liberação do hormônio foliculestimulante (FSH) e do hormônio luteinizante (LH) pela hipófise, que, em conjunto, são designados como gonadotrofinas. Esses hormônios são responsáveis pela maturação folicular e a ovulação, bem como pela produção associada de hormônios gonadais femininos.
Qual seria a relação desses hormônios com a maturação folicular e o processo de ovulação?
Precisamos relembrar a regulação hormonal durante o ciclo ovariano, também denominado ciclo menstrual feminino.
O FSH liberado pela hipófise no organismo promove o crescimento e a maturação dos folículos ovarianos, que induzem o aumento da síntese de estradiol, também conhecido por 17β-estradiol, o estrogênio mais potente produzido e secretado pelo ovário. Ele é responsável pelo aparecimento das características femininas, pelo crescimento endometrial e aumento da permeabilidade do muco cervical. No entanto, a partir de um mecanismo de retroalimentação negativa da adeno-hipófise, quando a concentração plasmática de estradiol se encontra elevada, a liberação de FSH é inibida. Assim, imediatamente antes da ovulação, quando o folículo quase maduro está produzindo alta concentração de estradiol, há intensa secreção transitória de LH e FSH, que culmina na ovulação (este mecanismo ainda não está totalmente elucidado).
Retroalimentação negativa
A regulação por retroalimentação negativa é essencial para garantir o equilíbrio do meio interno (homeostase). Ela induz a uma resposta inibitória que se opõe ao estímulo que promoveu o desequilíbrio de determinada sinalização. Também pode ser definida como feedback negativo.

Após a ovulação, o folículo rompido dá origem ao corpo amarelo. >>> Este libera progesterona em resposta ao LH.
A progesterona promove, então, o desenvolvimento de um endométrio secretor, que pode acomodar após a implementação o embrião em formação.
O suprimento sanguíneo também cresce para fornecer quantidades aumentadas de nutrientes, se houver gravidez.
Os elevados níveis de progesterona que são liberados durante a segunda metade do ciclo menstrual (a fase lútea) inibem a produção de gonadotropinas e, assim, evitam ovulações adicionais, além de diminuir a permeabilidade do muco cervical.
É importante destacarmos que os folículos não rompidos continuam liberando estradiol sob influência do FSH. Após duas semanas, a produção de progesterona e estradiol declinam, causando desprendimento da camada endometrial secretora, dando início à menstruação. Esse processo está ilustrado na Figura 10.




Caso ocorra a implementação do embrião (nidação), a progesterona continua sendo secretada, mantendo o endométrio em estado favorável para a continuidade da gestação. Assim, impede-se a menstruação. Além disso, a implantação no revestimento uterino induz o blastocisto a secretar gonadotropina coriônica humana (hCG), responsável por estimular o corpo lúteo a permanecer viável e continuar secretando progesterona.
Após a menstruação, na ausência de estrógeno e progesterona, há aumento da produção de FSH e LH, estimulando o desenvolvimento de novos folículos ovarianos e o início de outro ciclo ovariano.
Também é importante destacarmos que, após a síntese dos hormônios apresentados anteriormente, eles são liberados e difundem-se no plasma. Neste, podem se ligar fortemente a proteínas plasmáticas, e apenas a fração não ligada é capaz de difundir-se para as células, ligar-se ao receptor intracelular (ERα e ERβ), serem translocados até o núcleo, culminando em aumento ou inibição da transcrição de genes específicos, gerando, desse modo, os efeitos fisiológicos.
Após entendermos o papel de cada hormônio em nosso organismo, podemos começar a discutir as principais propriedades dos fármacos que atuam nesses processos fisiológicos. Alguns pacientes podem apresentar ruptura do eixo hipotálamo-hipófise-gonadal e precisam de reposição hormonal, como ocorre em mulheres na menopausa, ou podem apresentar um crescimento inapropriado de tecido dependente de estrógeno, como o desenvolvimento de câncer de mama, endometriose ou hiperplasia endometrial.
As principais indicações clínicas que estudaremos incluem contracepção hormonal, reposição hormonal pós-menopausa, queixas menstruais e climatéricas graves (instabilidade vasomotora), infertilidade, endometriose e até mesmo tratamento do câncer de mama.
O mecanismo de retroalimentação negativa, que discutimos no início deste módulo, será a base para o entendimento do mecanismo de ação da maioria dos fármacos.
Exemplo
Os contraceptivos orais (fármacos anticoncepcionais) são compostos por hormônios sintéticos que induzirão esse mecanismo de retroalimentação, alterando a síntese de outros hormônios, e inibindo a ovulação. Portanto, foi observado que a administração pulsátil de GnRH exógeno (simulando a produção fisiológica) estimula a liberação de gonadotrofinas, enquanto sua administração contínua inibe a liberação de LH e FSH e, consequentemente, bloqueia a função da célula-alvo.
De forma geral, embora os hormônios naturais endógenos possam ser absorvidos por via oral, eles não são utilizados por esta via, porque sofrem rápida metabolização hepática pré-sistêmica após sua absorção. No organismo, os metabólitos hidrossolúveis do estradiol e da progesterona, denominados estriol (também presente como estrona –metabólito ativo) e pregnandiol, respectivamente, são facilmente excretados no rim. No entanto, foram realizadas alterações na estrutura química desses hormônios, aumentando sua estabilidade após administração oral (metabolização mais lenta).

Assim, atualmente, existem diversas indicações clínicas para seu uso (hormônios sintéticos), que começaremos a discutir a seguir.
Estrógenos
Existem inúmeras preparações disponíveis comercialmente de estrógenos e eles se apresentam como:
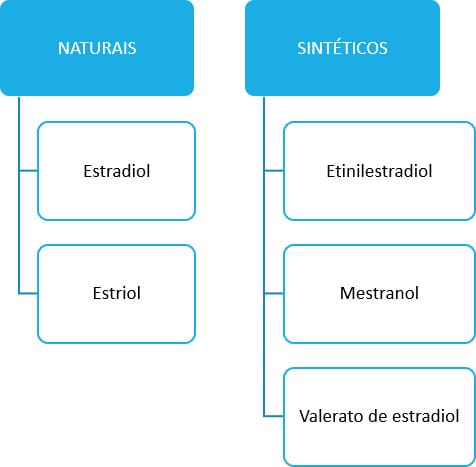
Dentre eles, o etinilestradiol pode ser administrado por via oral por apresentar boa disponibilidade e ser metabolicamente estável, ou seja, ele consegue ultrapassar o fígado e mimetizar o estradiol nos receptores de estrogênio. Este fármaco também é o principal componente estrogênico dos contraceptivos orais, que discutiremos com mais detalhes a seguir. Os outros medicamentos são pró-fármacos, ou seja, precisam ser metabolizados para se tornarem ativos.

Estrutura química do Etinilestradiol

Estrutura química do Mestranol

Estrutura química do Valerato de Estradiol
O mestranol é rapidamente desmetilado a etinilestradiol, já o valerato de estradiol é rapidamente clivado a estradiol e ácido valérico.
Esses análogos sintéticos são lipossolúveis e armazenados no tecido adiposo, de onde podem ser liberados lentamente. Desse modo, apresentam ação prolongada e maior potência quando comparados com os estrogênios naturais.
Os efeitos dos estrógenos exógenos estão relacionados diretamente ao estágio de maturação sexual quando administrado.
Exemplo
Antes da puberdade, os estrógenos podem estimular o desenvolvimento das características sexuais secundárias e acelerar o crescimento. Em pacientes com distúrbios de insuficiência ovariana, eles podem ser utilizados como terapia de reposição hormonal. Nos adultos com amenorreia, os estrógenos associados aos progestógenos podem induzir um ciclo artificial e serem usados para contracepção.
Amenorreia
Falta de menstruação nas mulheres em idade fértil.
Também existem os estrogênios conjugados (sulfatados), utilizados por via oral, mas apresentam fraca atividade e são mais indicados para o tratamento de reposição hormonal e durante ou após a menopausa, prevenindo os sintomas climatéricos (ondas de calor, ressecamento da vagina e diminuição da libido).
Além desses análogos disponíveis para utilização via oral, existem disponíveis as preparações de depósito, que são administradas por meio da injeção intramuscular. Os ésteres de estradiol são preparados em soluções oleosas, garantindo velocidade de liberação e duração prolongada do efeito. No organismo, quando o éster é hidrolisado, há liberação de estradiol. A utilização dessas substâncias em adesivo transdérmico também é realizada e apresenta boa penetrabilidade transcutânea do estradiol.
A administração de estrogênios sintéticos contínua, durante o início do ciclo ovariano, permite bloquear a produção de FSH. Com a redução do estímulo do FSH no folículo, a maturação dele é prejudicada, impedindo a ovulação. Com a administração do estrogênio durante a primeira metade do ciclo, as alterações fisiológicas no endométrio, bem como os outros efeitos no organismo, acontecem normalmente. A interrupção dos hormônios (período de pausa) é seguida da menstruação.
No entanto, a utilização de anticoncepcionais com predomínio estrogênico ou isolados não é aprovada, pois foi observada associação com o desenvolvimento de risco de câncer endometrial. Portanto, eles são sempre administrados em combinação com os progestogênios, conforme abordaremos a seguir.
Os efeitos adversos dos estrógenos incluem sensibilidade na mama, náusea, vômito, anorexia e edema. A administração de estrógenos é contraindicada durante a gravidez, podendo causar anomalias genitais no recém-nascido.
Progestógenos
As preparações dos progestogênios sintéticos para administração oral são derivadas da etiniltestosterona ou do acetato de 17α-hidroxiprogesterona. Esses progestogênios são frequentemente utilizados para contracepção e podem ser utilizados isolados ou em combinação com os estrógenos. A seguir, abordaremos alguns exemplos da terapia contraceptiva com os progestogênios isolados.
Etiniltestosterona
Por exemplo: noretisterona, norgestrel, etinodiol, linestrenol, desogestrel, gestodeno.
Acetato de 17α-hidroxiprogesterona
Por exemplo: hidroxiprogesterona, acetato de clormadinona ou acetato de ciproterona.

Tal como apresentado nas preparações de estrogênio, existem as formulações de depósito para uso intramuscular ou subcutâneo das preparações de progestogênios (administrados a cada três meses), compostos principalmente por caproato 17-α-hidroxiprogesterona e acetato de medroxiprogesterona. Eles também atuam na primeira metade do ciclo ovariano, bloqueando a produção de FSH. Muitas mulheres que utilizam essas formulações apresentam amenorreia, e o retorno da fertilidade pode demorar vários meses após sua descontinuação. Por isso, o fármaco não deve ser continuado por mais de dois anos.

A contracepção também pode ser realizada com administração contínua de doses baixas de progestogênios, que alteram a frequência dos pulsos de GnRH e promovem alterações na função cervical e endometrial, como, por exemplo espessando o muco cervical, dificultando o deslocamento dos espermatozoides. Atualmente, os fármacos que contêm apenas noretisterona, norgestrel ou desogestrel são denominados “minipílulas”. São pouco utilizados devido à necessidade de ingestão sempre na mesma hora do dia e estão associados a menor taxa de sucesso (menos eficazes do que as formas combinadas) e a frequentes ciclos menstruais irregulares. No entanto, eles possuem grande importância clínica, pois são indicados para mulheres que estão amamentando, para pacientes intolerantes ou que possuem contraindicações ao uso do estrogênio.
Você já ouviu falar na “pílula do dia seguinte”? Refere-se à administração de uma progesterona sintética em dosagem elevada (1,5 mg) em até três dias após o coito sem proteção. O mecanismo de ação do impedimento da gestação não foi totalmente esclarecido, mas acredita-se que, quando o levonorgestrel atua antes da ovulação, ele impede o aumento do LH induzido pela ovulação, além de produzir alterações endometriais que impossibilitam a implementação no útero. Os efeitos adversos mais frequentes deste fármaco são náuseas e êmeses.

Vamos conhecer mais sobre alguns progestógenos.
A mifepristona é um antagonista dos receptores da progesterona e impede a manutenção do endométrio no início da gestação, atuando, consequentemente, como fármaco abortivo. A mifepristona é comumente administrada em associação com misoprostol, análogo de prostaglandina, que discutiremos no módulo 4. O principal efeito adverso é sangramento uterino significativo e possibilidade de aborto incompleto. A coadministração de misoprostol pode causar náuseas e vômitos.
O modulador seletivo do receptor de progesterona acetato de ulipristal, conhecido como pílula dos cinco dias, também é utilizado para contracepção de emergência, se utilizado em até cinco dias após o coito sem proteção. Este fármaco ocupa os receptores da progesterona, impedindo que este hormônio promova o seu efeito, inibindo a ovulação e, consequentemente, a gravidez.
Especificamente para a indicação de contracepção, também há a possibilidade da utilização de um implante na camada dérmica, como o etonogestrel, que proporciona contracepção por cerca de três anos, e o efeito é totalmente reversível quando o implante é removido. O principal efeito adverso do implante é sangramento menstrual irregular e cefaleia.
O dispositivo intrauterino de liberação de levonorgestrel também constitui um método altamente eficaz de contracepção por três a cinco anos, dependendo do sistema.
Até aqui, abordamos somente a utilização isolada tanto dos estrogênios quanto dos progestogênios. No entanto, a maioria dos contraceptivos estão disponíveis em associação. Dessa forma, como funcionam esses anticoncepcionais?
Contraceptivos combinados (associação de estrogênios e progestogênios sintéticos)
Esses contraceptivos orais combinados constituem o método mais potente de suprimir a secreção de GnRH, LH e FSH e o desenvolvimento folicular, inibindo a ovulação, além de outros mecanismos secundários, como prevenção de crescimento endometrial (indicados para o tratamento da endometriose), redução de condições favoráveis à nidação, diminuição de permeabilidade do muco cervical aos espermatozoides, que, associados, garantem eficácia contraceptiva superior a 95%, evitando a concepção.
Muitas mulheres apresentam períodos menstruais menos intensos quando fazem uso de contraceptivos orais combinados, além de apresentarem redução dos sintomas menstruais. Anemia por deficiência de ferro e tensão pré-menstrual também são reduzidas.
Atualmente, existem disponíveis diferentes tipos de associação para utilização por via oral, mas sempre com base na associação de um estrogênio com um progestogênio.
A associação mais utilizada e clássica é denominada monofásica, terapia que é baseada na administração constante desses hormônios (mesma dosagem) de 21 a 24 dias, seguidos de quatro a sete dias de placebo (etapa em que ocorre a descamação do endométrio e consequente sangramento).

Totalizando um regime de 28 dias, aproximadamente.
Além dessa associação monofásica, existem protocolos bifásicos, trifásicos e até mesmo de quatro fases, quando há tentativa de mimetizar o ciclo natural feminino, no qual há uma dose constante de estrogênio com doses crescentes de progestogênios administradas a cada sete dias. Entretanto, não existem diferenças significativas relacionadas a efeitos adversos ou à eficácia clínica entre essas terapias e a monofásica.

Estrutura química do Etinilestradiol
Estrutura química do Mestranol
Os estrogênios sintéticos mais utilizados nesses fármacos são o etinilestradiol e o mestranol. Atualmente, prefere-se utilizar a menor dose de etinilestradiol, pois acredita-se induzir a menor risco de trombose venosa profunda nas pacientes.

Estrutura química do Noretisterona

Estrutura química do Levonorgestrel

Estrutura química do Desogestrel

Estrutura química do Norgestimato

Estrutura química da Drospirenona
Os progestogênios sintéticos mais comuns, que atuam como agonistas dos receptores da progesterona, são noretisterona, acetato de noretisterona, levonorgestrel, desogestrel, norgestimato e drospirenona.
Atualmente, com o objetivo de reduzir a biotransformação dos fármacos, existem alternativas ao uso do contraceptivo oral. Por exemplo, eles podem ser utilizados por via intravenosa (injeções), transdérmica (adesivo, gel e emulsão tópica ou spray), contendo a associação de etinilestradiol e norelgestromina, e via intravaginal (óvulo, creme ou anéis vaginais), contendo associação de etinilestradiol e etonogestre. Todas essas alternativas são utilizadas por um momento, geralmente três semanas, e, em seguida, têm seu uso interrompido por sete dias, para ocorrer a menstruação.

Os efeitos adversos ocasionados pela utilização dessas combinações são raros, mas podem ocorrer. Por exemplo, ganho de peso, edema, náusea, tontura, depressão, mudanças na pele (pigmentação), entre outros.
Atenção
Após o uso prolongado desses fármacos, também foi observado maior risco, embora sejam eventos raros, de desenvolver câncer de mama, tumores hepáticos benignos, alterações coronarianas, infarto do miocárdio, acidente vascular encefálico e tromboembolismo. No entanto, o risco de câncer ovariano e endometrial é reduzido, bem como fibrose uterina e cistos funcionais do ovário. Foi relatada diminuição na incidência de fraturas ósseas, mas eles não são indicados para este fim, devido à relação risco-benefício desfavorável.
O aumento de incidência de tromboembolismo está relacionado ao uso do estrogênio em particular, com aumento da coagulação sanguínea, mas também a alguns progestogênios específicos, como gestodeno (ver imagem) e desogestrel. No entanto, existem fatores predisponentes, como história familiar, tabagismo, obesidade e idade, que podem favorecer esse aumento. De modo geral, o consenso é de que anticoncepcionais orais apresentam mais efeitos clínicos benéficos do que prejudiciais.

Conforme discutimos, as duas classes de contraceptivos orais amplamente utilizadas são combinações de estrógeno-progestógeno, além de existirem contraceptivos somente com progestógeno isolado.
Vamos abordar agora alguns fármacos não esteroidais que se ligam ao receptor de estrogênio e exercem efeito agonista ou antagonista seletivo, dependendo do tipo de tecido, denominados moduladores seletivos de receptores de estrogênio (MSREs), que possuem diversas indicações clínicas.
Moduladores seletivos de receptores de estrogênio (MSRE)
O hormônio estradiol é agonista dos receptores de estrogênio. No entanto, existem vários fármacos que mimetizam essa ligação e outros que atuam como antagonistas desses receptores.
Dentre eles, o clomifeno é utilizado por via oral para tratamento da infertilidade feminina com ciclos anovulatórios. Ao antagonizar o receptor de estrogênio no hipotálamo e na hipófise, a retroalimentação negativa da gonadotropina pelo estradiol é suprimida, o que promove aumento da liberação de GnRH, gonadotrofinas e indução da maturação dos folículos ovarianos. O clomifeno é indicado para mulheres com insuficiência na maturação folicular ou no tratamento da síndrome do ovário policístico. Como a sua utilização é curta, os efeitos crônicos não são considerados. É comum o nascimento de gêmeos, mas a gravidez múltipla é incomum. Seu principal efeito adverso consiste na capacidade de induzir o crescimento de múltiplos folículos, resultando em aumento de tamanho do ovário.
Anovulatórios
Quando um óvulo não é liberado do ovário durante um ciclo menstrual.


O tamoxifeno também é um antagonista dos receptores de estrógeno no tecido mamário, porém agonista parcial no endométrio e nos ossos. É atualmente aprovado para o tratamento e a prevenção do câncer de mama, bloqueando o crescimento das células tumorais. Além disso, pode ser utilizado em mulheres com queixas do climatério. De forma preocupante, como apresenta efeito agonista estrogênico no endométrio, pode impulsionar o crescimento das células tumorais sensíveis ao estrogênio e tem sido observada maior incidência de câncer. Assim, a sua utilização deve ser realizada com cautela, durante um período que não deve ultrapassar cinco anos.
Já o raloxifeno atua como agonista estrogênico nos ossos (diminui a reabsorção óssea), mas apresenta atividade antagonista nos tecidos mamário e endometrial. Foi aprovado para tratamento e profilaxia de osteoporose pós-menopausa, mas atualmente existem outras recomendações mais benéficas, como o uso de alendronato de sódio. O raloxifeno também é aprovado para uso em prevenção de câncer de mama.

Todos os MSREs são rapidamente absorvidos após administração oral e sofrem extensa biotransformação por isoenzimas do citocromo P450. Portanto, deve-se atentar para a utilização desses fármacos em pacientes com polimorfismos genéticos ou em associação a outros fármacos. Apresentam ciclo entero-hepático e são excretados principalmente através da bile para as fezes. Os efeitos adversos mais frequentes são queixas climatéricas e náuseas.
Inibidores da aromatase
O mecanismo de ação dos inibidores seletivos da aromatase baseia-se na inibição da produção de estrogênio, visto que eles bloqueiam a enzima aromatase, responsável pela conversão de andrógenos em estrógenos.

Estrutura química do Anastrozol

Estrutura química do Letrozol
O anastrozol e o letrozol são inibidores competitivos da aromatase.

Estrutura química do Exemestano

Estrutura química do Formestano
O exemestano e o formestano ligam-se de modo irreversível à aromatase.
Esses fármacos são utilizados principalmente no tratamento do câncer de mama avançado, quando o tumor é sensível ao estrogênio e a paciente encontra-se na menopausa. Como o estrógeno constitui importante regulador da densidade óssea, mulheres em uso contínuo de inibidores da aromatase possuem risco elevado de desenvolver fraturas osteoporóticas.
Recentemente, tem sido discutida a possibilidade de ligação de estrógenos e progestógenos em outros tecidos. Portanto, existe uma nova área de pesquisa farmacológica em desenvolvimento, em que pesquisadores continuam desenvolvendo agonistas e antagonistas desses receptores, com ações seletivas em tecidos específicos, como os MSRE, o que, consequentemente, aumentará sua utilização clínica no futuro.
No vídeo a seguir, abordaremos as características dos esteroides anabólicos e a utilização dessa classe de substância para fins estéticos.
Verificando o aprendizado
ATENÇÃO!
Para desbloquear o próximo módulo, é necessário que você responda corretamente a uma das seguintes questões:
O conteúdo ainda não acabou.
Clique aqui e retorne para saber como desbloquear.
MÓDULO 4
Identificar as principais propriedades farmacológicas, os mecanismos de ação, as indicações clínicas, os efeitos colaterais e as contraindicações dos ocitócicos
Conceitos
Neste módulo, abordaremos os fármacos que atuam estimulando o útero, denominados ocitócicos. Conforme abordado no módulo 3, as respostas fisiológicas do útero variam durante o ciclo menstrual e até mesmo durante a gravidez a partir da regulação hormonal. A definição de ocitócicos vem do grego e significa “parto rápido”, o que os correlaciona com os efeitos clínicos desses fármacos.
A regulação da contração do útero é realizada pelas células miometriais, que atuam como marcapasso no útero, sendo a atividade eletrofisiológica regulada pelos hormônios sexuais. Os estrogênios aumentam a contração uterina. A progesterona, por sua vez, reduz a contração. Além disso, o sistema nervoso autônomo desempenha um papel importante nas células miometriais. Enquanto o sistema parassimpático estimula a contração, o sistema simpático pode promover o relaxamento uterino após ligação da epinefrina aos receptores β2-adrenérgicos, ou estimular a contração após ligação da norepinefrina nos receptores α.

Durante o ciclo ovariano, foi observado que o útero humano não gravídico se contrai (com menor intensidade) durante a fase folicular, que termina com a liberação de LH. Já durante a fase lútea e na menstruação, o útero se contrai com maior intensidade. No início da gestação, os movimentos uterinos são reduzidos, devido à liberação de progesterona e hipertrofia das células miometriais, que suprimem as contrações. No entanto, ao final da gestação, as contrações recomeçam, aumentam em força e frequência, e tornam-se coordenadas durante o parto.
Diversos fármacos são utilizados para estimular o útero gravídico e são importantes na obstetrícia. Entre eles, destacam-se:
- Ocitocina.
- Ergometrina.
- Prostaglandina.
Começaremos a abordar as principais propriedades farmacológicas destes fármacos. A utilização clínica com mais detalhes será discutida em um caso clínico, no Explore +.
O principal efeito farmacológico induzido por esses fármacos é a contração uterina. Para que ela ocorra, é necessário aumento dos níveis de cálcio na célula muscular, resultado de ativação da sinalização intracelular, acoplada à proteína G. Além disso, os ocitócicos podem estimular a formação de miosina de cadeia leve, por meio de uma quinase (MLCK), induzindo à contração.
Todos esses fármacos podem promover inúmeros efeitos adversos graves e irreversíveis à mãe e ao feto. Dessa forma, os sinais vitais, tanto maternos quanto fetais, devem ser monitorados enquanto esses fármacos forem utilizados.

Ocitocina
A ocitocina é um decapeptídeo endógeno sintetizado no hipotálamo, armazenada na neuro-hipófise e, posteriormente, secretada de forma pulsátil para a corrente sanguínea, em resposta a sinais fisiológicos específicos, regulando a atividade miometrial. Os estímulos para a secreção de ocitocina incluem estímulos sensoriais, que se originam da dilatação do colo do útero e da vagina, além da sucção da mama. Ela provoca contrações do músculo liso do útero coordenadas e regulares, seguidas, cada uma delas, de relaxamento.

A liberação de estrógeno durante o ciclo ovariano estimula a secreção de ocitocina e promove a síntese e a expressão dos receptores de ocitocina. Assim, o útero gravídico torna-se muito sensível a este hormônio. A ocitocina também induz a produção de ácido araquidônico pela decídua, o que aumenta a produção local de prostaglandinas (suas propriedades serão discutidas a seguir), potencializando o efeito contrátil da ocitocina.
Decídua
Camada funcional da mucosa uterina onde a placenta está implantada e que se hipertrofia durante a gestação. Sua função é conferir proteção ao embrião.
A ocitocina sintética, idêntica ao hormônio natural, pode ser utilizada clinicamente com maior frequência por injeções intravenosas ou até mesmo por intramusculares, e apresenta meia-vida curta (12-15 min). A depuração da ocitocina é renal e hepática, decorrente da ação da enzima ocitocinase, enzima circulante produzida pela placenta e com alta atividade durante a gestação.
As principais indicações terapêuticas da ocitocina exógena em obstetrícia incluem induzir ou estender o trabalho de parto que não está em progressão, principalmente quando o músculo uterino não funciona corretamente. Também pode ser utilizada na profilaxia e/ou no tratamento de hemorragia pós-parto.
Vamos conhecer o efeito da ocitocina durante e após a gestação.
A ocitocina é o fármaco de escolha para indução de trabalho de parto. Para esta finalidade, ela é administrada por infusão intravenosa em baixas concentrações, promovendo contrações coordenadas regulares. Além desses efeitos, utilizada em baixas doses e de uso prolongado, favorece o amolecimento do colo e promove aumento do número de seus receptores no miométrio. Nessa fase, a ocitocina induz poucas contrações, permitindo o descanso na fase inicial do trabalho de parto. Discutiremos a existência de outros fármacos mais convenientes para o amolecimento do colo, uma vez que a ocitocina demora muitas horas (12-18h) para produzir o afinamento e o encurtamento do colo uterino.
Logo após o amolecimento do colo (aumento da dilatação do colo uterino), infunde-se a ocitocina em doses progressivamente maiores para realizar a indução do trabalho de parto. Conforme já abordamos, a amplitude e a frequência de contrações estão relacionadas à dose, ou seja, em doses menores, o útero relaxa completamente entre as contrações, e, em doses maiores, há aumento das contrações e relaxamento incompleto entre elas. A utilização de concentrações elevadas pode promover contrações sustentáveis, que interferem no baixo fluxo sanguíneo através da placenta, podendo induzir a sofrimento fetal, havendo risco de levar a óbito. Portanto, as contrações uterinas e a frequência cardíaca fetal devem permanecer sob monitoramento contínuo.
Outro papel fisiológico muito importante que a ocitocina desempenha é o de promover a ejeção do leite. A estimulação da mama por meio da sucção ou de manipulação mecânica induz à secreção de ocitocina, que provoca contração das células mioepiteliais da glândula mamária. Essa ação força a saída do leite presente nos canais alveolares para os grandes seios coletores, ficando disponível para o lactante.
Neste sentido, outra indicação terapêutica aprovada na Anvisa é a administração intranasal de ocitocina sob a forma de spray, com o objetivo de estimular a secreção de leite (ejeção de leite) em mulheres com dificuldade para amamentar ou extrair o leite, na prevenção e no tratamento do ingurgitamento lácteo das mamas e na prevenção da mastite.
Ingurgitamento lácteo
Produção exagerada de leite e seu acúmulo nas mamas, popularmente conhecido como leite empedrado.
Após a absorção pela mucosa nasal, o efeito sobre a mama ocorre em menos de cinco minutos. Em casos de administração de volume excessivo de solução spray nasal, a ocitocina é rapidamente inativada no trato digestivo pelas enzimas proteolíticas. O uso da ocitocina sob a forma de spray é contraindicado durante a gravidez, podendo ocorrer contrações uterinas e indução ao aborto.
É preciso atentar para a administração intravenosa, pois a ocitocina também pode apresentar outros efeitos indesejáveis, como ação diurética e efeito vasodilatador – efeitos que são desencadeados por apresentar estrutura química muito semelhante à da vasopressina (hormônio antidiurético).
Os principais efeitos adversos da utilização de ocitocina para a mãe incluem hipotensão dose-dependente com taquicardia reflexa, ruptura uterina, hipotonia uterina pós-parto (tônus muscular enfraquecido), deslocamento de placenta, retenção de líquido. Em casos em que a ingestão de água esteja diminuída, pode promover hiponatremia.
Assim, a utilização de ocitocina em pacientes com doenças cardíacas e renais, ou com pré-eclâmpsia, deve ser realizada com cautela. Pré-eclâmpsia é uma condição patológica associada a episódios de hipertensão arterial, após a 20ª semana de gravidez, associada a proteinúria significativa (presença de proteína na urina). Se não tratada, em alguns casos, as pacientes podem apresentar edema e convulsões, caracterizados como eclâmpsia.
Outro aspecto a respeito do qual se deve ter cautela durante a indução do trabalho de parto são as possíveis complicações de hiperestimulação uterina, que incluem traumatismo à mãe ou ao feto em consequência de passagem forçada através de um colo do útero não completamente dilatado, além do comprometimento da oxigenação fetal por diminuição da perfusão uterina. A administração de ocitocina pode resultar em bradicardia e hiperbilirrubinemia neonatal.

Sua utilização é contraindicada para pacientes com hipersensibilidade conhecida do fármaco, desproporção cefalopélvica e posição ou apresentação fetal desfavoráveis. Além disso, não deve ser usada para induzir o trabalho de parto, quando o parto vaginal for contraindicado.
Desproporção cefalopélvica
(do grego-latim cephalos, cabeça)
É um termo obstétrico para quando a pelve materna não é larga o suficiente para permitir a passagem da cabeça do bebê.
Existem evidências após estudos em animais que apontam a ocitocina como um importante regulador do SNC e dos sistemas autônomos, associados à modulação de ansiedade e medo, e na regulação de respostas neuroendócrinas e cardiovascular. Em humanos, no entanto, essa associação ainda não foi estabelecida.
Esse vídeo abordará outros efeitos fisiológicos da ocitocina, que vão além daqueles relacionados à obstetrícia e lactação.
Análogos sintéticos da ergometrina
A ergometrina (alcaloide) foi isolada em 1935 a partir de uma variedade de substâncias ativas derivadas do fungo esporão do centeio ou ergot (Claviceps purpurea). Os relatos na literatura evidenciam que o envenenamento por ergot na Antiguidade era frequentemente associado ao aborto. Assim, foi denominada como um princípio ativo ocitócico do ergot.

Nos casos em que a ocitocina não for eficaz, a ergometrina pode ser utilizada clinicamente para tratar a hemorragia pós-parto, pois, como todos os ocitócicos, seu principal efeito farmacológico é promover a contração do útero humano com aumento do tônus basal. No entanto, esse efeito é mais pronunciado se o útero estiver inapropriadamente relaxado, quando ela promove forte contração, reduzindo o sangramento uterino. Em uma condição em que o útero já se encontra contraído em seu estado normal após a expulsão do feto, a ergometrina tem relativamente pouco efeito.

Estrutura química da Metilergornovina

Estrutura química da Metilergometrina
Os análogos sintéticos da ergometrina, como a metilergonovina ou metilergometrina, são facilmente absorvidos pelo trato gastrointestinal e podem ser administrados por via oral, além de poderem ser utilizados por via intramuscular ou intravenosa. Os efeitos são observados em três a cinco minutos após a administração por via intramuscular, e seu efeito dura de três a seis horas.
Os alcaloides derivados do ergot apresentam várias ações. São estruturalmente semelhantes à noradrenalina, à dopamina e à serotonina, podendo apresentar efeitos agonistas ou antagonistas sobre os receptores α-adrenérgicos (principal efeito ocitócico), de dopamina e 5-hidroxitripamina. Portanto, outro efeito farmacológico característico da utilização da ergometrina é a ação vasoconstritora moderada. Os principais efeitos adversos são:
- Episódios de vômitos.
- Elevação da pressão sanguínea associada a náusea.
- Visão turva.
- Cefaleia.
- Angina.
A ergometrina pode ser administrada em associação com a ocitocina durante o trabalho de parto. Além disso, pode ser utilizada antes da cirurgia para controle de sangramento devido a aborto incompleto. É contraindicada para pacientes com hipertensão não controlada.
Análogos das prostaglandinas
As prostaglandinas são produzidas fisiologicamente derivadas de ácido araquidônico e afetam a maioria dos tecidos. Embora apresentem meia-vida plasmática curta, estão associadas ao controle de vários processos fisiológicos, incluindo fluxo sanguíneo, diâmetro das vias aéreas, ovulação, tônus do músculo liso uterino, e são as principais mediadoras e moduladoras da resposta inflamatória, que constituem alvo de inúmeros fármacos. Não há nenhum mecanismo de ação comum a todas as prostaglandinas.
O endométrio e o miométrio possuem a capacidade de síntese de determinadas prostaglandinas, particularmente na segunda fase do ciclo menstrual. Entre elas, encontram-se a PGF2α, PGE2 e PGI1 (também chamada de prostaciclina), promovendo efeito vasodilatador. Além desta propriedade, as PGE2 e PGF2α promovem contrações coordenadas do útero gravídico, enquanto produzem relaxamento do colo uterino. A sensibilidade do músculo uterino às prostaglandinas aumenta durante a gestação.
As prostaglandinas sintéticas têm sido empregadas para indução do trabalho de parto desde 1970, quando se tornaram disponíveis por via vaginal. As mais utilizadas, que mimetizam os efeitos das produzidas fisiologicamente, são a dinoprostona (PGE2), carboprosta (15-metil PGF2α) e a gemeprosta ou misoprostol (análogos da PGI1). O objetivo delas é aumentar a dilatação do colo uterino antes do início do trabalho de parto com ocitocina ou em situações em que a mulher já iniciou o trabalho de parto, mas o colo ainda está pouco dilatado.

A dinoprostona pode ser administrada por via intravaginal, em gel, anel ou em comprimidos, para o amadurecimento do colo do útero e na indução do trabalho de parto. Também por via extra amniótica, em solução, utilizada tardiamente, no segundo trimestre da gravidez, para induzir o aborto terapêutico. Foi observado que a utilização da PGE2 sintética aumentou a chance de parto vaginal em 24 horas, mas também aumentou a hiperestimulação uterina, com alteração dos batimentos fetais, e não reduziu a taxa de parto cesáreo.
Via extra amniótica
Injeção do medicamento entre o córion (membrana exterior do óvulo) e o âmnio (membrana fina que reveste o interior onde se encontra o feto).

A carboprosta é administrada por injeção intramuscular profunda, e é utilizada para induzir abortamento no segundo trimestre e para o controle da hemorragia puerperal que não responda a tratamentos tradicionais.

O misoprostol está aprovado pela Anvisa para o manejo de aborto espontâneo ou induzido somente em uso obstétrico no ambiente hospitalar, no entanto ele é frequentemente vendido no mercado clandestino como abortivo, o que pode induzir inúmeros efeitos deletérios para a gestante e o feto. É administrado por via oral, sublingual ou intravaginal, como óvulos vaginais, induzindo o amadurecimento do colo do útero. Quando é combinado com o fármaco mifepristona (RU486), antagonista dos receptores da progesterona, o misoprostol é altamente eficaz para a interrupção precoce da gravidez. O misoprostol também pode ser utilizado para prevenção e tratamento de hemorragia pós-parto.

Pela via vaginal, o misoprostol promove menos efeitos colaterais, pois a primeira etapa do metabolismo hepático é evitada, mantendo-se os níveis plasmáticos elevados por um período mais prolongado. No fígado, o misoprostol sofre desesterificação, transformando-se no principal metabólito ativo, o ácido misoprostol. Este exerce ação direta nos receptores das prostaglandinas. O misoprostol deve ser interrompido pelo menos três horas antes do início da terapia com ocitocina.
Os principais efeitos adversos que ocorrem quando esses fármacos são utilizados incluem: dor uterina (ocasionado por hiperestimulação uterina), ruptura uterina (rara), náusea, vômito, diarreia e cólica devido a aumento da taxa de contração do músculo longitudinal do TGI. Pode ocorrer flebite no local da administração intravenosa. Além disso, por promoverem constrição venosa superficial e contração do músculo liso dos brônquios, a carboprosta é contraindicada em pacientes asmáticos. A dinoprostona não deve ser usada em mulheres com história de asma, glaucoma ou infarto miocárdico.
A produção aumentada de prostaglandinas pelo útero pode prejudicar a homeostasia, assim como causar vasodilatação. Esses fenômenos estão associados a duas das principais doenças da menstruação, a dismenorreia (menstruação dolorosa) e a menorragia (perda excessiva de sangue). No sentido de inibir a produção excessiva dessas prostaglandinas, alguns anti-inflamatórios não esteroidais (AINES), como o ácido mefenâmico, podem ser utilizados para inibir a biossíntese das prostaglandinas no útero e tratar a menorragia e a dismenorreia. Alguns inibidores da ciclo-oxigenase também podem retardar o trabalho de parto, impedindo a produção das prostaglandinas aqui discutidas.
Verificando o aprendizado
ATENÇÃO!
Para desbloquear o próximo módulo, é necessário que você responda corretamente a uma das seguintes questões:
O conteúdo ainda não acabou.
Clique aqui e retorne para saber como desbloquear.
Conclusão
Considerações Finais
Ao final deste material, podemos afirmar que aprendemos as principais propriedades farmacológicas das terapias atualmente utilizadas para diversos distúrbios endócrinos frequentes em nossa população, como diabetes, hipotireoidismo e hipertireoidismo.
Além disso, abordamos os principais mecanismos de ação, as características farmacocinéticas, os eventos adversos e, entre outras peculiaridades, os da terapia contraceptiva e da utilização dos ocitócicos. Não podemos, porém, esquecer que novos estudos e novas pesquisas são realizados continuamente e que este conhecimento é diariamente renovado.
Podcast
CONQUISTAS
Você atingiu os seguintes objetivos:
Descreveu as propriedades farmacológicas da insulina e dos hipoglicemiantes orais, os mecanismos de ação, as principais indicações clínicas, os efeitos colaterais e as principais contraindicações
Descreveu a fisiologia dos hormônios da tireoide e as propriedades farmacológicas dos fármacos antitireoidianos, os mecanismos de ação, as principais indicações clínicas, os efeitos colaterais e as contraindicações
Reconheceu as propriedades farmacológicas dos estrógenos e progestógenos, os mecanismos de ação, as principais indicações clínicas, os efeitos colaterais e as contraindicações
Identificou as principais propriedades farmacológicas, os mecanismos de ação, as indicações clínicas, os efeitos colaterais e as contraindicações dos ocitócicos
O tromboembolismo é caracterizado pela formação de coágulos (trombos) no interior de vasos sanguíneos superficiais ou profundos, principalmente dos membros inferiores. Essa condição patológica é denominada trombose venosa (quando ocorre em veias profundas, é chamada de Trombose Venosa Profunda – TVP). Os trombos podem ser deslocados e alcançar os pulmões, causando embolia pulmonar. Uma vez que as pílulas anticoncepcionais são um fator de risco relacionado à TVP, o tromboembolismo é um efeito adverso que preocupa muitas mulheres que utilizam esses fármacos como anticoncepcionais.