Descrição
Estrutura, nomenclatura, propriedades físicas, reatividade, sínteses, fontes e uso de compostos oxigenados não carbonilados e aminas.
PROPÓSITO
Apresentar noções básicas sobre estrutura, nomenclatura, propriedades, métodos de obtenção e reações de compostos oxigenados não carbonilados e aminas, dando ênfase aos mecanismos das reações.
OBJETIVOS
Módulo 1
Reconhecer as características químicas, físicas e reatividade dos compostos oxigenados não carbonilados, bem como suas nomenclaturas
Módulo 2
Identificar os princípios mecanísticos gerais e as principais reações de compostos oxigenados não carbonilados
Módulo 3
Identificar as características químicas, físicas e reatividade das aminas, assim como suas nomenclaturas
Módulo 4
Reconhecer os princípios mecanísticos gerais e as principais reações com aminas
Introdução
Os compostos oxigenados não carbonilados e as aminas estão entre os mais abundantes na natureza, podendo ser encontrados em biomoléculas ativas e recursos naturais. Além disso, possuem muitas aplicações industriais e farmacêuticas.
Neste tema, você terá a oportunidade de reconhecer as principais reações que envolvem os álcoois, fenóis, éteres e as aminas, assim como descrever os princípios mecanísticos de cada tipo de reação. Abordaremos também alguns exemplos de reações e suas aplicações no nosso cotidiano.
MÓDULO 1
Reconhecer as características químicas, físicas e reatividade dos compostos oxigenados não carbonilados, bem como suas nomenclaturas
QUÍMICA DOS COMPOSTOS OXIGENADOS NÃO CARBONILADOS: ÁLCOOIS, ÉTERES, EPÓXIDOS E FENÓIS
Álcoois e fenóis ocorrem em abundância na natureza e possuem muitas aplicações industriais e farmacêuticas.

O metanol, por exemplo, embora seja tóxico aos seres humanos em pequenas doses, é um dos mais importantes produtos químicos industriais, sendo utilizado como solvente de tintas e vernizes, como material de partida para a produção do formaldeído e do ácido acético, na síntese de insumos farmacêuticos, na produção de biodiesel e como combustível de motores à explosão, como aviões a jato e carros de corrida.

Em contrapartida, a produção de etanol por meio da fermentação de grãos e açúcares é conhecida há cerca de 9.000 anos, tendo a sua purificação pela destilação iniciada no século XII. O etanol é o álcool de todas as bebidas alcoólicas e, apesar de dar impressão de ser um estimulante, ele é hipnótico (induz ao sono), diminuindo a atividade na parte superior do cérebro.

O fenol é encontrado no alcatrão de hulha (tipo de carvão) e possui como principais aplicações a antisséptica e a germicida, pois consegue coagular as proteínas dos organismos e das bactérias. Entretanto, por possuírem essa ação desinfectante, os fenóis também podem ser tóxicos e cáusticos.
Já entre os éteres, podemos citar o dietílico e o dietilenoglicol.
O éter dietílico talvez seja a substância mais familiar entre os éteres. Em 1842, ele foi empregado como anestésico cirúrgico na Geórgia e logo depois foi introduzido no uso cirúrgico em um hospital em Boston, entretanto é um líquido altamente volátil que atualmente entrou em desuso por ser inflamável (SOLOMONS e FRYHLE, 2001).
O dietilenoglicol (DEG) apresenta propriedade anticongelante, sendo muito utilizado nas indústrias farmacêutica e cosmética e em motores de carros, mas também pode ser tóxico aos seres humanos. No início de 2020, o dietilenoglicol foi a principal causa de intoxicação em diversas pessoas que consumiram cerveja artesanal produzida no estado de Minas Geras que estava contaminada pela substância. Algumas delas morreram e outras tiveram a saúde comprometida devido à síndrome nefroneural causada pelo DEG. Acredita-se que o uso inadequado da substância para refrigeração dos tanques com um possível vazamento tenha causado a contaminação da cerveja.
Síndrome nefroneural
Danos causados aos rins e ao sistema nervoso.
Além dos usos citados, os éteres são utilizados como solventes de óleos, gorduras, resinas e na fabricação de seda artificial.
Estruturas e propriedades dos álcoois e fenóis
Os álcoois são derivados orgânicos que possuem o grupo hidroxila ligado a um átomo de carbono saturado. O átomo de carbono pode ser de um grupo alquila simples, alquenila, alquinila ou o átomo de carbono saturado pode estar ligado a um anel benzênico. Além disso, eles podem ser classificados como primários, secundários ou terciários, dependendo do número de grupos orgânicos ligados ao carbono que possui a hidroxila.

Já os compostos que apresentam um grupo hidroxila ligado diretamente a um anel benzênico são classificados como fenóis. Nesse caso, o fenol é o nome específico de um composto, hidroxibenzeno, e o nome geral para uma família de compostos derivados do hidroxibenzeno.

Essas classes de compostos apresentam praticamente a mesma geometria em torno do átomo de oxigênio, possuindo um ângulo da ligação R-O-H de aproximadamente 109° e o átomo de oxigênio com hibridização sp3.

A presença de hidroxila nessas classes de compostos permite que eles possuam uma capacidade de formar ligações de hidrogênio, fazendo com que os álcoois e fenóis estejam associados entre si e, consequentemente, tenham um ponto de ebulição maior do que seus isômeros funcionais que não fazem esse tipo de interação (os éteres, por exemplo).
O átomo de hidrogênio do –OH polarizado positivamente de uma molécula é atraído pelo par de elétrons no átomo de oxigênio eletronegativo de outra molécula, resultando em uma força fraca que mantém as moléculas unidas.
Atenção
Sendo assim, para vencer essas atrações intermoleculares, é necessário maior temperatura para que as moléculas entrem no estado de vapor.
Podemos observar na figura abaixo que os álcoois, devido às suas ligações de hidrogênio, possuem pontos de ebulição bem maiores quando comparados com os hidrocarbonetos de massas moleculares próximas.

Outra propriedade dos álcoois e fenóis, proporcionada pela habilidade em formar ligações de hidrogênio, é a sua solubilidade em água. Entretanto, a solubilidade dos álcoois diminuirá gradualmente à medida que a cadeia de hidrocarboneto da molécula aumentar.
Tabela 1: Propriedades físicas dos álcoois e fenóis.
|
Composto |
Nome |
p.f. (°C) |
p.e. (°C) |
Solubilidade em água |
|
Álcoois monoidroxilados |
||||
|
CH3CH2OH |
Etanol |
-117 |
78,3 |
∞ |
|
CH3CH2CH2OH |
Propanol |
-126 |
97,2 |
∞ |
|
CH3CH2CH2CH2OH |
Butanol |
-90 |
117,7 |
8,3 |
|
(CH3)3COH |
Terc-butanol |
25 |
82,5 |
∞ |
|
CH3CH(CH3)CH2OH |
Isobutanol |
-108 |
108,0 |
10,0 |
|
CH3CH2CH2CH2CH2OH |
Pentanol |
-78,5 |
138,0 |
2,4 |
|
CH3CH2CH2CH2CH2CH2OH |
Hexanol |
-52 |
156,5 |
0,6 |
|
CH2=CHCH2OH |
2-propenol |
-129 |
97 |
∞ |
|
Dióis |
||||
|
CH2OHCH2OH |
Etileno glicol |
-12,6 |
197 |
∞ |
|
CH3CHOHCH2OH |
Propileno glicol |
-59 |
187 |
∞ |
|
Fenóis |
||||
|
C6H5-OH |
Fenol |
43 |
182 |
9,3 |
|
o-CH3C6H5-OH |
2-metilfenol |
30 |
191 |
2,5 |
|
m-CH3C6H5-OH |
3-metilfenol |
11 |
201 |
2,6 |
|
p-CH3C6H5-OH |
4-metilfenol |
35,5 |
201 |
2,3 |
|
o-ClC6H5-OH |
2-clorofenol |
8 |
176 |
2,8 |
|
m-ClC6H5-OH |
3-clorofenol |
33 |
214 |
2,6 |
|
p-ClC6H5-OH |
4-clorofenol |
43 |
220 |
2,7 |
Nomenclatura dos álcoois e fenóis
Na nomenclatura substitutiva IUPAC, um nome pode ter até quatro características:
Localizador
Prefixos
Composto principal
Sufixo
Veja o exemplo abaixo com o composto 4-metil-pentan-2-ol.

Os álcoois simples são sistematicamente nomeados substituindo-se o sufixo -o da maior cadeia hidrocarbônica que contém a hidroxila pelo sufixo -ol.
Atenção
A cadeia hidrocarbônica deve ser numerada de maneira que o número menor fique mais próximo ao carbono ligado à hidroxila.
As posições do grupo hidroxila e dos outros substituintes (prefixos) devem ser indicadas como localizadores, conforme Figura 4.
Conforme recomendações da nomenclatura IUPAC, o localizador da hidroxila deve ser colocado imediatamente antes do sufixo e os demais localizadores devem ser dispostos no nome do composto em ordem alfabética, conforme Figura 5.

Apesar dos nomes sistemáticos ou substitutivos IUPAC, os álcoois mais simples e abundantes apresentam nomes comuns, que também são aprovados pela IUPAC (Figura 6).

Já os álcoois que possuem duas hidroxilas não terão o sufixo –o substituído por –ol, e sim a adição do sufixo –diol, pois neste caso não há possibilidade de ocorrência de duas vogais adjacentes, como nos álcoois com apenas uma hidroxila. Entenda melhor esse conceito observando os exemplos na figura abaixo.

Por fim, os fenóis substituídos podem ser nomeados com o sufixo –fenol ao final do nome da cadeia principal.
Atenção
Entretanto, os compostos que possuem um grupo hidroxila ligado a um anel benzenoide são chamados de naftóis e fenatróis, embora, sejam quimicamente similares aos fenóis.
Os metilfenóis e benzenodióis também apresentam nomes comuns, chamados de cresóis e catecol, resorcinol e hidroquinona, respectivamente.

Estruturas e propriedades dos éteres e epóxidos
Os éteres são derivados orgânicos que possuem dois grupos hidrocarbônicos ligados ao mesmo átomo de oxigênio. Os grupos podem ser alquila, alquenila, vinila, alquinila ou arila e o átomo de oxigênio pode fazer parte de uma cadeia linear ou de um anel (Figura 9).

Assim como os álcoois, as ligações R-O-R dos éteres possuem um ângulo de ligação próximo ao tetraédrico e o oxigênio com hibridização sp3. As temperaturas de ebulição são comparáveis às dos hidrocarbonetos de mesmo peso molecular, entretanto as temperaturas serão mais baixas que as dos álcoois comparáveis.
Atenção
Os éteres, contudo, podem realizar ligações de hidrogênio com a água, fazendo com que sua solubilidade seja semelhante aos álcoois de mesmo peso molecular.

Dentro da família dos éteres, nós temos os éteres cíclicos que possuem o mesmo comportamento que os ésteres de cadeia linear, com exceção dos epóxidos.
Epóxidos
Os epóxidos são éteres cíclicos com anéis de três membros que, devido à tensão do anel, possuem uma reatividade única. Dentre os epóxidos, o óxido de etileno é o mais simples e utilizado como intermediário na fabricação do etileno glicol e de polímeros de poliéster.

Além dos éteres já citados anteriormente, os éteres cíclicos, dioxano e tetraidrofurano são muito utilizados na indústria como solventes em razão da sua inércia.
Inércia
O sentido de inércia neste contexto se deve ao fato dessas substâncias atuarem apenas como solventes, sem que interfiram no curso de reações orgânicas ou provoquem modificações estruturais nos compostos solubilizados por eles.
Nomenclatura dos éteres e epóxidos
Os éteres simples são geralmente nomeados por termos comuns, denominados radicofuncionais. Os dois grupos ligados ao oxigênio são listados em ordem alfabética, com o sufixo –ico adicionado ao último, precedidos pela palavra éter.
Atenção
Entretanto, para compostos mais complexos ou com mais de uma ligação de éter, os nomes substitutivos da IUPAC devem ser usados.
Nesse caso, os éteres são denominados como alcoxialcanos, alcoxialcenos e alcoxiarenos (RO- é o grupo alcoxi). A nomenclatura será o prefixo que indica o número de carbonos do menor radical, mais –oxi e o nome do hidrocarboneto correspondente ao maior radical, conforme mostrado nos exemplos da figura abaixo.

Os éteres cíclicos podem ser denominados de diferentes maneiras. A primeira é a nomenclatura de substituição, onde o éter cíclico é relacionado ao sistema de anel do hidrocarboneto correspondente, utilizando o prefixo oxa-. A segunda maneira é o radical referente à cadeia hidrocarbônica com o sufixo –eno, precedidos da palavra óxido (Figura 13). Já o sistema de um éter cíclico de três membros é chamado de oxirano ou epóxido, enquanto o éter cíclico de quatro membros é chamado de oxetano.

Reatividade química e acidez dos compostos oxigenados não carbonilados
Os fenóis, assim como a água, apresentam comportamento anfótero, ou seja, se comportam tanto como ácidos fracos quanto base fracas. Como bases, eles são reversivelmente protonados por ácidos fortes para produzir íons oxônio, ROH2+. Já como ácidos, eles se dissociam fracamente em solução aquosa, doando um protón para a água, formando H3O+ e um alcóxido ou fenóxido.
Saiba mais
Usualmente, é comum na química orgânica empregarmos a sigla “Ar” para representar a estrutura do anel benzênico. Assim, é comum o fenol ser simbolizado nos textos específicos da área como ArOH, por exemplo.
Álcoois simples como o metanol e o etanol são tão ácidos quanto a água, mas a substituição por grupamentos alquilas terá um efeito significativo na acidez dos álcoois, devido à solvatação do íon alcóxido que resulta da dissociação. Observe na tabela abaixo que álcoois mais impedidos estericamente são menos ácidos que os álcoois não impedidos, pois esses são favorecidos pela maior facilidade de solvatação pela água, estabilizando o átomo de oxigênio do respectivo íon alcóxido.
Tabela 2: Valores de pKa de alguns álcoois e fenóis.
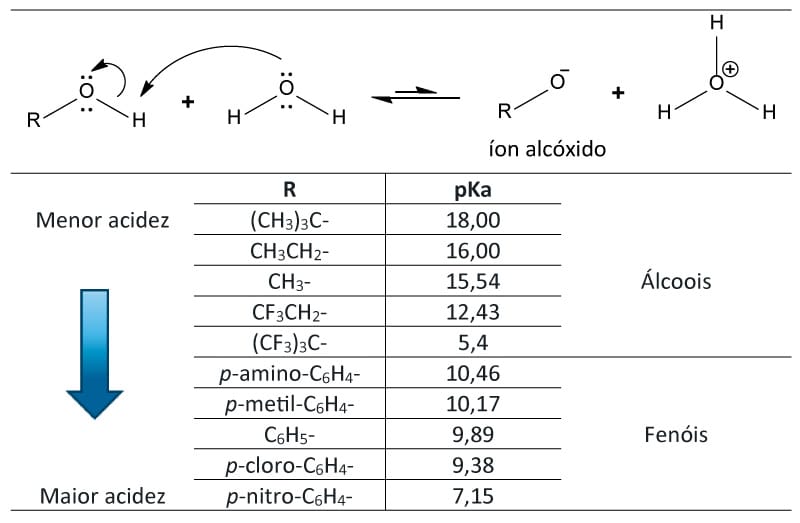
Ainda sobre a tabela, podemos observar que o efeito indutivo do substituinte também influenciará na acidez dos compostos. Note que os compostos que possuem halogênios como substituinte apresentam
um menor valor de pKa em relação aos seus correspondentes sem o substituinte.
No álcool nonafluoro-tert-butila, por exemplo, o caráter eletronegativo do flúor é o responsável por “puxar” elétrons da ligação C-F. O carbono, por sua vez, puxa elétrons do carbono ligado à hidroxila, que puxará elétrons do átomo de oxigênio. Consequentemente, a carga negativa é dispersa ao longo da molécula, estabilizando o alcóxido.
Atenção
Quanto maior o número de substituintes puxadores de elétrons (como o flúor, por exemplo) e o quão mais perto eles estiverem da ligação O–H, maior a acidez do composto e, consequentemente, menor será o pKa.
Os álcoois são frequentemente utilizados como solventes para reações orgânicas por apresentarem maior facilidade que a água em dissolver compostos orgânicos menos polares. O seu uso como solvente tem como vantagem a utilização dos íons alcóxido como base. Outra vantagem do uso desses compostos como solventes é a possibilidade de serem gerados íons alcóxido no meio reacional e estes atuarem como espécies básicas.
Exemplo
Podemos criar uma solução de etóxido de sódio em álcool etílico pela adição de hidreto de sódio (NaH) ao álcool. Entretanto, devemos utilizar um grande excesso de álcool etílico, pois queremos que, além de serem gerados os íons etóxido, o meio contenha muitas moléculas de álcool para que ele atue como o solvente.

Agora, para falarmos dos fenóis, vamos voltar à tabela 2 e utilizá-la como base para a nossa discussão. Note que os fenóis possuem valores de pKa menores que os álcoois, isso ocorre porque o íon fenóxido é estabilizado por ressonância.
Conforme podemos observar na figura abaixo, a deslocalização da carga negativa sobre as posições orto e para do anel aromático ocasiona o aumento da estabilidade do fenóxido em relação ao fenol não dissociado.

Assim como nos álcoois, os substituintes influenciarão na reatividade dos fenóis. Os fenóis com substituintes retiradores de elétrons são frequentemente mais ácidos que o fenol devido à capacidade de os substituintes distribuírem a carga negativa. Já os substituídos por doadores de elétrons serão menos ácidos que o fenol, uma vez que ocasionam a concentração da carga negativa.

Já os éteres são relativamente estáveis e não reativos, em geral, só sofrem basicamente reações de clivagem por ácidos e, devido a essa propriedade, são muito usados como solventes para outros compostos orgânicos. Em contrapartida, os epóxidos se comportam de maneira diferente dos éteres de cadeia linear devido à tensão do anel de três membros, proporcionando uma maior reatividade a estes compostos.
As fontes naturais dos compostos oxigenados não carbonilados
Como mencionamos, os compostos oxigenados não carbonilados ocorrem em abundância na natureza. Neste tópico, observaremos algumas biomoléculas conhecidas e de fontes naturais.

Nossa primeira molécula é o eugenol ou 4-alil-2-metoxifenol, composto aromático, principal componente do óleo de cravo e encontrado na canela, mirra e nas sassafrás. Possui propriedades antimicrobianas, anti-inflamatórias, espasmolíticas, antipiréticas, antissépticas e anestésicas, sendo muito utilizado no tratamento de dores de dente. A adição de amina ao metileugenol leva ao 3,4 dimetoxianfetamina, ou 3,4-DMA, uma anfetamina alucinógena largamente consumida em festas “raves”.

Os urushióis são agentes vesicantes, constituintes alergênicos, encontrados, principalmente, no veneno do carvalho e da hera. A toxicidade desses compostos é atribuída à presença de compostos fenólicos e catecólicos ou à mistura de lipídeos fenólicos.

A vanilina ou 3-metoxi-4-hidroxibenzaldeído, muito conhecida na culinária, é o principal componente da essência de baunilha. É um composto orgânico cristalino de cor branca com os grupos funcionais fenol, éter e aldeído, e suas principais aplicações são na indústria de alimentos, bebidas e produtos farmacêutico como agente aromatizante.

O timol ou 2-isopropil-5-metil-fenol é um composto orgânico pertencente ao grupo dos terpenos, podendo ser extraído do tomilho. Possui propriedades carminativas, antiespamódicas, expectorantes e anti-inflamatória. Há relatos de que muitas civilizações antigas já conheciam sobre as propriedades antisséptica do tomilho e do timol. Os antigos egípcios, por exemplo, já o utilizavam no interior de sarcófagos para maximizar a conservação do ambiente, evitando a proliferação de germes e bactérias. Na Idade Média, os ramos de tomilho eram utilizados pelos juízes nos tribunais para afastar infecções.
O estradiol é um hormônio sexual feminino e esteroide. É importante na regulação do ciclo estral e do ciclo menstrual, além de ser essencial para o desenvolvimento e manutenção dos tecidos reprodutivos femininos e de ter efeitos importantes em muitos outros tecidos, incluindo o ósseo.
O anisol ou metoxibenzeno é um éter aromático, líquido, volátil, com odor agradável originário da semente de anis e muito utilizado na indústria de cosméticos e de higiene.
Outro éter importante encontrado na natureza é o eucaliptol ou 1,8-cineol, um monoterpeno incolor encontrado na composição de alguns óleos essenciais, como o de alecrim, eucalipto, sálvia e manjericão. Há estudos que demonstram que o eucaliptol possui propriedades anti-hipertensiva, antiasmática, analgésica e vasodilatadora.

Nomenclatura e propriedades dos compostos oxigenados não carbonilados
Verificando o aprendizado
ATENÇÃO!
Para desbloquear o próximo módulo, é necessário que você responda corretamente a uma das seguintes questões:
O conteúdo ainda não acabou.
Clique aqui e retorne para saber como desbloquear.
MÓDULO 2
Identificar os princípios mecanísticos gerais e as principais reações de compostos oxigenados não carbonilados
Neste módulo, nós iremos abordar diversas formas de preparar álcoois, éteres e epóxidos, além de mostrar as reações de proteção e oxidação de álcoois e fenóis.
SÍNTESE DOS ÁLCOOIS
Os álcoois podem ser preparados a partir de vários tipos de compostos, como alcenos, haletos de alquila, cetonas, ésteres e aldeídos.

A obtenção de álcoois a partir da hidratação de alcenos ocorre na presença de água e um catalisador ácido.

A adição segue a regra de Markovnikov, produzindo álcoois secundários e terciários, exceto no caso do eteno. Entretanto, é uma reação que geralmente resulta em rendimentos baixos no laboratório e em que ocorre rearranjos com frequência, limitando o seu uso.
Já a oximercuração-desmercuração e hidroboração-oxidação são dois métodos mais comumente utilizados nos laboratórios. Na oximercuração-desmercuração, os alcenos reagem com acetato mercúrico em THF/água para formar compostos hidroxialquilmercúricos para que assim possam ser reduzidos a álcoois com boroidreto de sódio.
Regra de Markovnikov
Na adição de um haleto de hidrogênio a um alceno, ou na hidratação de um alceno, o hidrogênio do haleto ou da água liga-se ao átomo de carbono mais hidrogenado da dupla ligação, ou seja, ao carbono menos substituído.
Rearranjos
Rearranjos de metila ou hidreto ocorrem sempre que um carbocátion menos estável puder se transformar em um outro mais estável.

Na primeira etapa, oximercuração, o íon eletrofílico HgOAc+ aceita um par de elétrons do alceno para formar um carbocátion com ponte de mercúrio, fazendo com que a carga positiva do carbocátion seja compartilhada entre o átomo de carbono secundário e o átomo de mercúrio. Em seguida, uma molécula de água ataca o carbono que carrega a carga positiva parcial e, por fim, uma reação ácido-base transfere um próton a uma outra molécula de água (ou íon acetato), produzindo o composto hidroxialquil mercúrico e íon hidrônio.
Observe que o método de oximercuração-desmercuração suplementa a hidratação, proporcionando um método adicional para a adição de Markovnikov, porém, sem ser atrapalhada pelos rearranjos.
O mecanismo para a segunda etapa, desmercuração, onde ocorre a substituição do grupo acetoximercúrico pelo hidrogênio ainda é pouco compreendida, mas acredita-se que envolve formação de radicais.
Na hidroboração-oxidação, há a possibilidade de adicionar o –H e –OH a um alceno de uma forma anti-Markovnikov, resultando em uma adição sin global.

A hidroboração ocorre com tratamento do alceno com solução de THF:BH3. O hidreto de boro adiciona sucessivamente às ligações duplas de três moléculas do alceno, formando o tripropilborano. Em cada etapa de adição, o átomo de boro se liga ao átomo de carbono menos substituído da ligação dupla e um átomo de hidrogênio é transferido do átomo de boro ao outro átomo do carbono da ligação dupla.
Atenção
A regiosseletividade anti-Markovnikov possivelmente é resultado de fatores estéricos, ou seja, o grupo volumoso que contém o boro pode se aproximar mais facilmente do átomo de carbono menos substituído.

No mecanismo proposto, inicia-se com a doação de elétrons π da ligação dupla ao orbital p vazio do BH3 (complexo π), em seguida, forma-se um estado de transição de quatro centros cíclicos com a adição do átomo de boro ao átomo de carbono menos substituídos. Observe na figura 23 que as linhas tracejadas representam as ligações que são parcialmente formadas ou parcialmente rompidas. Por fim, o estado de transição se transforma em um alquilborano. As demais ligações B-H podem passar por adições semelhantes, levando ao trialquilborano.
A segunda etapa é a oxidação dos triaquilboranos formados. O átomo de boro aceita um par de elétrons do íon hidroperóxido, formando um intermediário instável que terá um grupo alquila migrando do boro para o átomo de oxigênio adjacente quando um íon hidróxido se afasta. A migração do grupo alquila (com retenção da configuração) irá ocorrer até que todos eles se liguem aos átomos de oxigênio, dando origem a um borato de trialquila.

Por fim, o éster formado sofrerá hidrólise básica para produzir três moléculas do álcool e um íon borato.
Álcoois a partir da redução de compostos de carbonila
A redução de um composto de carbonila para a preparação de um álcool é um dos métodos gerais mais presentes em laboratórios, onde todos os compostos de carbonila podem ser reduzidos, incluindo aldeídos, cetonas, ácidos carboxílicos e ésteres.
As reduções de aldeídos e cetonas podem ser realizadas com diferentes reagentes, entretanto, o boroidreto de sódio é o mais utilizado por questões de segurança e facilidade de manuseio.
Aldeídos
São reduzidos a álcoois primários.
Cetonas
São reduzidas a álcoois secundários.

Já as reduções de ácidos carboxílicos e ésteres formam álcoois primários, porém, como não são tão rápidas como as reduções de aldeídos e cetonas, é necessária a utilização de um agente redutor mais reativo, como o LiAlH4.

Álcoois a partir da reação de compostos de carbonila com reagentes de Grignard
Nesta reação, os reagentes de Grignard irão agir como nucleófilos, reagindo com os compostos de carbonila para gerar os álcoois primários a partir de formaldeído, secundários a partir de aldeídos e terciários a partir de cetonas. Os ácidos carboxílicos não geram produtos de adição com os reagentes de Grignard, pois o hidrogênio ácido do grupo carboxílico reage com o reagente de Grignard básico, dando origem a um hidrocarboneto e um sal de magnésio do ácido. As reações com ésteres formam álcoois terciários nos quais dois dos substituintes ligados ao carbono do grupo hidroxila são oriundos do reagente de Grignard.

OXIDAÇÃO DE ÁLCOOIS E FENÓIS
A reação de oxidação de álcoois é uma das mais importante dentre as reações com álcoois, sendo capaz de produzir aldeídos ou ácidos carboxílicos a partir de álcoois primários e cetonas a partir de álcool secundários. Já os álcoois terciários não reagem com a maioria dos agentes oxidantes.

Os álcoois primários são oxidados tanto a aldeídos como ácidos carboxílicos, dependendo do reagente e das condições empregadas. A oxidação de álcoois primários em aldeídos dificilmente consegue parar em aldeído, sendo necessários agentes de oxidação especiais. Um dos mais utilizados é o clorocromato de piridínio, formado quando o CrO3 é dissolvido em ácido clorídrico e depois tratado com piridina. A maioria dos outros agentes oxidantes, como o trióxido de cromo ou permanganato de potássio, é capaz de oxidar os álcoois primários diretamente a ácidos carboxílicos.

Os álcoois secundários são oxidados facilmente para formar cetonas. Dentre os vários agentes oxidantes baseados no cromo (VI) disponíveis, o mais utilizado é o ácido crômico preparado pela adição do óxido de cromo (VI) ou dicromato de sódio ao ácido sulfúrico aquoso.

A primeira etapa do mecanismo de oxidação do álcool pelo ácido crômico é a formação de um éster cromato do álcool, produto instável e não isolado. Posteriormente, o éter cromato transfere um próton a uma base, normalmente a água, e simultaneamente elimina um íon HCrO3- para a formação de uma acetona.
Já a oxidação em fenóis não ocorre da mesma maneira que nos álcoois, pois não há um átomo de hidrogênio no carbono que contém o grupo hidroxila. Sendo assim, a reação do fenol com um agente oxidante forte, nitrodissulfonato de potássio (KSO3)2NO, produz uma quinona (ou 2,5-cicloexadieno-1,4-diona). A reação ocorre em condições brandas através de um mecanismo radicalar.

PROTEÇÃO DE ÁLCOOIS
Durante a síntese de moléculas complexas pode ocorrer de um grupo funcional em uma molécula interferir na reação de um segundo grupo funcional presente na mesma molécula. Quando isso ocorre, às vezes, é possível resolver o problema protegendo o grupo funcional interferente, seguindo três etapas:
A formação do éter trimetilsilílico é o método mais comum para proteção de álcoois, onde o clorotrimetilsilano reage com o álcool na presença de uma base, geralmente trietilamina.

O éter formado não possui hidrogênios ácidos, sendo pouco reativo e, consequentemente, não reage com agentes oxidantes, agentes redutores ou reagentes de Grignard, reage apenas com ácido aquoso ou com íon fluoreto para regenerar o álcool.
Exemplo
No exemplo a seguir é possível verificar a utilização do grupo protetor para realizar a reação de Grignard em um halo-álcool.

SÍNTESE DE ÉSTERES E EPÓXIDOS
O éter dietílico e outros éteres simétricos são preparados industrialmente pela reação de desidratação de álcoois catalisada por ácido sulfúrico. A desidratação ocorre a uma temperatura mais baixa que a desidratação em um alceno (aprox. 140°C). A formação do éter ocorre por um mecanismo SN2 com uma molécula de álcool agindo como o nucleófilo e com uma outra molécula protonada de álcool agindo como o substrato.
Entretanto, esse método é limitado à utilização de álcoois primários, pois o uso de álcoois secundários e terciários levam à formação de alcenos através de um mecanismo E1.

Para éteres assimétricos, o caminho mais conveniente é a síntese de Williamson, na qual um íon alcóxido reage com um haleto de alquila primário ou tosilato em uma reação SN2. Como qualquer reação SN2, a síntese de Williamson possui algumas limitações, tais como a utilização de haletos de alquila, sulfonato ou sulfato primários, pois em substratos mais impedidos pode ocorrer competição com a reação de eliminação E2.
Atenção
Sendo assim, os éteres assimétricos são melhor preparados pela reação do íon alcóxido mais impedido e um haleto menos impedido.

Na síntese de éteres, podemos, também, empregar a alcoximercuriação de alcenos.
Neste caso, um alceno é tratado com um álcool na presença de acetato mercúrico ou, ainda melhor, trifluoroacetato de mercúrico. Em seguida, a desmercuriação com NaBH4 forma o éter, tendo como resultado a adição de Markovnikov do álcool a um alceno. O mecanismo de reação é similar àqueles demonstrados para o preparo de álcoois que vimos anteriormente.

Por fim, os epóxidos são preparados pelo tratamento de um alceno com um peroxiácido, processo chamado de epoxidação.
Nessa reação, o perácido transfere um átomo de oxigênio ao alceno em um mecanismo cíclico de uma única etapa, formando um epóxido e um ácido carboxílico, através de uma adição sin do oxigênio ao alceno.

Síntese de álcoois e éteres
Verificando o aprendizado
ATENÇÃO!
Para desbloquear o próximo módulo, é necessário que você responda corretamente a uma das seguintes questões:
O conteúdo ainda não acabou.
Clique aqui e retorne para saber como desbloquear.
MÓDULO 3
Identificar as características químicas, físicas e reatividade das aminas, assim como suas nomenclaturas
INTRODUÇÃO À QUÍMICA DAS AMINAS
As aminas são compostos orgânicos largamente encontrados em organismos vivos e utilizados em diversos tipos de indústria, sendo junto com os compostos carbonilados os quimicamente mais ricos.
Na natureza, ela pode ser encontrada:
Como a nicotina presente no tabaco, a Indigotina presente no anil, o corante natural encontrado na Indigofera anil, a cafeína encontrada no café, o guaraná e mate, e a cocaína, estimulante encontrado nos arbustos da planta da coca na América do Sul que já foi usada em medicamentos e em bebidas.
Atualmente, seu maior consumo é através de drogas ilícitas que causam dependência química.
É possível encontrar trimetilamina, responsável pelo odor característico dos peixes, histrionicotoxina, neurotoxina extremamente tóxica, presente em sapos colombianos, aminoácidos, blocos fundamentais a partir dos quais todas as proteínas são formadas e as bases de aminas cíclicas, constituintes dos ácidos nucleicos.
Temos a presença das aminas na produção de sabão, condicionadores para cabelo, produção de corantes sintéticos para uso em alimentos e medicamentos e na vulcanização da borracha, processo em que se adiciona enxofre à borracha natural para torná-la mais resistente e flexível. Na indústria farmacêutica, as aminas são amplamente utilizadas na produção de medicamentos e estão muito presentes nas classes de anfetaminas, antidepressivos, ansiolíticos e vitaminas.
Estrutura e propriedades das aminas
As aminas são compostos orgânicos nitrogenados derivados da amônia e podem ser alquil-substituídas ou aril-substituídas. A ligação das alquilaminas é semelhante àquela na molécula de amônia. O átomo de nitrogênio possui hibridização sp3, onde os três grupos alquila (ou hidrogênios) ocupam os vértices do tetraedro e o orbital sp3, contendo o par de elétrons não compartilhado, está dirigido para o outro vértice.

Os ângulos de ligação C-N-C são aqueles esperados para uma estrutura tetraédrica, ou seja, próximos a 109°. A estrutura tetraédrica pode proporcionar uma amina com três substituintes diferentes no átomo de nitrogênio, caracterizando-a como quiral, ou seja, existirão duas formas enatioméricas da amina terciária. Entretanto, diferentemente dos compostos quirais de carbono, a resolução das aminas enantioméricas é normalmente impossível de ser realizada, pois os enantiômeros se interconvertem rapidamente através de inversão piramidal. Como a barreira para inversão é de aproximadamente 25 kJ mol-1, ela é suficientemente baixa para ocorrer rapidamente à temperatura ambiente.
As aminas são substâncias moderadamente polares, podendo realizar ligações de hidrogênio entre elas e a água e com pontos de ebulição mais altos do que aqueles com alcanos, mas geralmente são mais baixos do que aqueles dos álcoois de massa molecular comparável. As aminas terciárias, como só podem realizar ligações de hidrogênio com as moléculas de água, geralmente, apresentam pontos de ebulição mais baixos que as aminas primárias e secundárias de massa comparável. Além disso, todas as aminas com menos de cinco átomos de carbono são solúveis em água.
Saiba mais
Outra característica marcante das aminas é o odor. As aminas de baixa massa molecular como a trietilamina possuem odor característicos de peixe, enquanto as diaminas, como a cadaverina (1,5-pentanodiamina) e a putrescina (1,2-butandiamina), possuem odores oriundos da decomposição de cadáveres.
Nomenclatura das aminas
As aminas podem ser classificadas como aminas primárias, secundárias e terciárias a depender do número de substituintes orgânicos.
Atenção
No caso das aminas, os substituintes estão ligados diretamente ao nitrogênio, diferentemente dos álcoois em que os substituintes estão ligados ao carbono ligado à hidroxila.

Existem também os compostos contendo o átomo de nitrogênio ligado a quatro grupos, denominados sais de amônio quaternários. Nesse caso, o nitrogênio deve ter uma carga formal positiva (Figura 40).

Na nomenclatura IUPAC, as aminas recebem os nomes adicionando-se o sufixo –amina ao nome da cadeia ou sistema cíclico ao qual o NH2 está ligado com a substituição do final –o. Para as aminas secundárias ou terciárias, utilizamos o localizador N para designar os substituintes ligados a um átomo de nitrogênio.
Já na nomenclatura comum, muitas aminas recebem os nomes como alquilaminas/arilaminas. Nas aminas secundárias e terciárias, designamos os grupos orgânicos individualmente se eles são diferentes ou utilizamos prefixos di- ou tri- se eles são iguais. Para substituintes diferentes, o maior grupo alquila deverá ser considerado como nome principal, e os outros grupos alquila serão considerados como N-substituintes da cadeia principal.

No sistema IUPAC, o substituinte –NH2 é chamado grupo amino, utilizado frequentemente para dar nomes às aminas contendo um grupo OH ou um grupo CO2H.

As aminas heterocíclicas são compostos nos quais o átomo de nitrogênio faz parte do anel.
Possuem nomenclatura comuns, onde cada anel heterocíclico recebe o seu próprio nome principal.
Na nomenclatura sistemática IUPAC, embora pouco utilizada, os prefixos aza-, diaza- e triaza- são utilizados para indicar que os átomos de carbono foram substituídos por átomos de nitrogênio no hidrocarboneto correspondente.

Reatividade e basicidade das aminas
As aminas possuem um par de elétrons isolados no nitrogênio que as tornam básicas e nucleofílicas, mais fortes que a água, os álcoois e os éteres, porém mais fracas que os íons hidróxidos e alcóxidos. Da mesma maneira que podemos medir a força de um ácido carboxílico por meio da constante de acidez, a força de uma base pode ser estabelecida pela constante de basicidade, ou seja, quanto maior o valor de Kb, menor o valor de pKb e mais forte a base.
Entretanto, os valores de Kb, na prática, não são muito utilizados, sendo assim, a maneira mais conveniente de se comparar a basicidade das aminas é comparar as constantes de acidez dos seus ácidos conjugados (íons alquilamínio correspondentes). O íon amínio de uma amina mais básica tem um Ka menor do que o íon amínio de uma amina menos básica.
Base fraca
pKa menores para o ácido conjugado → ácido mais forte.
Base forte
pKa maiores para o ácido conjugado → ácido mais fraco.
Tabela 3: Basicidade de algumas aminas.
| Nome | Estrutura | pKa do íon amínio |
|---|---|---|
|
Amônia |
NH3 |
9,26 |
|
Etilamina |
CH3CH2NH2 |
10,64 |
|
Dietilamina |
(CH3CH2)2NH |
10,98 |
|
Anilina |
C6H5-NH2 |
4,63 |
|
Piridina |
C5H5N |
5,25 |
|
Pirrol |
C4H5N |
0,4 |
Avaliando a tabela, podemos observar que os íons alquilamínio primários são menos ácidos do que o íon amônio, ou seja, as aminas primárias são mais básicas do que a amônia. Assim como, as aminas secundárias são mais básicas que as primárias. Isso acontece porque o grupo alquila tem habilidade de doar elétrons, estabilizando o íon alquilamínio que resulta da reação ácido-base através da dispersão da sua carga positiva.

Mas, e quanto às aminas terciárias? Elas não seriam as mais básicas?
Na fase gasosa, a basicidade das aminas se eleva com o aumento da substituição por grupos alquilas.
Entretanto, na fase aquosa, o comportamento não é o mesmo, pois os íons formados a partir das aminas primárias e secundárias são estabilizados pela solvatação através de ligação de hidrogênio com as moléculas de água.
Já nas aminas terciárias, esta solvatação já não é tão eficiente, pois os íons amínios correspondentes possuem apenas um hidrogênio para realizar ligação de hidrogênio com a água, enquanto os íons derivados das aminas primárias e secundárias possuem dois ou três, respectivamente. Em contrapartida, o efeito doador de elétrons faz com que a amina terciária seja mais básica do que a amônia.
Fase gasosa(CH3)3N > (CH3)2NH > CH3NH2 > NH3Solução aquosa(CH3)2NH > CH3NH2 > (CH3)3N > NH3 |
Em relação às arilaminas, normalmente, elas são menos básicas que as alquilaminas. Ao avaliarmos a tabela 2, podemos observar que os valores de pKa dos íons anilina e a p-toluidina, por exemplo, são menores que o do íon da cicloexilamina. Isso ocorre porque nas arilaminas o par de elétrons do nitrogênio está deslocalizado pela interação com o sistema de elétrons π do anel aromático, estando menos disponíveis para ligação com o H+.
Tabela 2: Basicidade de algumas aminas.
| Nome | Estrutura | pKa do íon amínio |
|---|---|---|
|
Amônia |
NH3 |
9,26 |
|
Etilamina |
CH3CH2NH2 |
10,64 |
|
Dietilamina |
(CH3CH2)2NH |
10,98 |
|
Anilina |
C6H5-NH2 |
4,63 |
|
Piridina |
C5H5N |
5,25 |
|
Pirrol |
C4H5N |
0,4 |

As estruturas 3, 4 e 5 são as responsáveis por contribuir com a deslocalização do par de elétrons, estabilizando a anilina. Outro ponto importante para a menor basicidade das aminas aromáticas é o efeito retirador de elétrons do grupo fenila, pois, uma vez que os átomos de carbono são hibridizados sp2, eles são mais eletronegativos e, consequentemente, mais retiradores de elétrons que os carbonos sp3 dos grupos alquilas.
As aminas e as biomoléculas
Os grupos aminas estão presentes em muitos compostos biológicos e utilizados na medicina, dentre eles, podemos citar as feniletilaminas. As 2-feniletilaminas são compostos com efeitos fisiológicos e psicológicos impressionantes.
Adrenalina
É um hormônio importante excretado na medula pela glândula adrenal. Quando o corpo de um animal se sente em perigo, a adrenalina é liberada na corrente sanguínea, causando um aumento da pressão sanguínea, um aumento da taxa de batimento cardíaco e uma abertura das passagens dos pulmões.
Dopamina
É um neurotransmissor da família das catecolaminas, também conhecidas como aminas biogênicas. Os neurotransmissores dessa família atuam no sistema nervoso central (SNC), modulando as funções de neurotransmissão relacionadas, por exemplo, ao movimento, às emoções, à atenção, ao aprendizado e ao sono. As normalidades no nível de dopamina no cérebro estão associadas com muitas desordens psiquiátricas, incluído a Doença de Parkinson.

Anfetamina
É uma droga sintética com propriedade estimulante e possui estrutura similar à adrenalina. As similaridades estruturais desses compostos devem estar relacionadas com os efeitos fisiológicos e psicológicos, pois muitos outros compostos com propriedades similares também são derivados da 2-feniletilamina. São drogas sintéticas que estimulam a atividade do SNC.
Edellano, em 1887, foi quem primeiro obteve essa substância em laboratório, que só foi utilizada em larga escala durante a Segunda Guerra Mundial para manter os soldados acordados e mais ativos no esforço de batalha. Foi observado, também, que as anfetaminas, além de serem eficazes para deixá-los mais atentos e confiantes, diminuíam a sensação de fome e fadiga. Entretanto, as autoridades médicas da Inglaterra notaram que o desempenho dos pilotos RAF sob efeito da droga ficava comprometido e o seu uso foi proibido. Após a confirmação da ação como inibidora do apetite, a anfetamina passou a ser usada nas dietas alimentares para perda de peso.
Atenção
Entretanto, infelizmente, existem usos inadequados da droga, como o rebite, utilizado pelos caminhoneiros para não dormir no volante, a bolinha que deixa o estudante “aceso” nas vésperas das provas e as “balinhas” ou comprimidos de ecstasy utilizados na noite pelos jovens.
Nomenclatura e propriedades das aminas
Verificando o aprendizado
ATENÇÃO!
Para desbloquear o próximo módulo, é necessário que você responda corretamente a uma das seguintes questões:
O conteúdo ainda não acabou.
Clique aqui e retorne para saber como desbloquear.
MÓDULO 4
Reconhecer os princípios mecanísticos gerais e as principais reações com aminas
Abordaremos neste módulo diversas maneiras de preparar aminas, além de mostrar as reações que envolvem o seu uso.
SÍNTESE DE AMINAS
A partir de substituição nucleofílica de haletos de alquila
Os sais de aminas podem ser preparados a partir de amônia/aminas e de haletos de alquila através de reações de substituição nucleofílica. O tratamento subsequente dos sais de amínio resultantes com uma base irá fornecer primárias, secundárias ou terciárias. Se for utilizada amônia, o resultado será uma amina primária; se for empregada uma amina primária, o resultado será uma amina secundária e assim sucessivamente; no caso da amina terciária, o produto formado será o sal de amônio quaternário.

Atenção
Mas, infelizmente, esse método é de aplicação muito limitada, pois não cessam após a primeira alquilação.
Uma vez que a amônia e as aminas primárias possuem a mesma reatividade, a substância monoalquilada formada incialmente pode sofrer reações múltiplas, formando uma mistura de produtos.
Um método muito mais eficaz para síntese de aminas primárias a partir de haleto de alquila é utilizar a síntese de azida. Neste caso:
Primeiro passo
Reagir o haleto de alquila com NaN3 (azida de sódio) a fim de gerar uma azida de alquila (R-N3) por uma reação de substituição nucleofílica. Uma vez que as azidas de alquila não são nucleofílicas, a reação não prossegue.
Segundo passo
A azida de alquila pode ser reduzida a uma amina primária com hidreto de lítio e alumínio. Vale destacar que as azidas de alquila de baixa massa molecular são explosivas e não devem ser isoladas, mas, sim, mantidas em solução.

A segunda alternativa para o preparo de aminas primárias é a Síntese de Gabriel que usa uma alquilação da ftalimida, que, por ser bastante ácida, pode ser convertida em ftalimida de potássio pelo hidróxido de potássio. O ânion de ftalimida formado por ser um nucleófilo forte irá reagir com o haleto de alquila através de um mecanismo SN2, fornecendo uma N-alquilftalimida.
Haleto de alquila
Restritos a haletos de metila, haletos de alquila primários e secundários.
Atenção
A N-alquilftalimida pode ser hidrolisada com ácido ou base aquosa (hidrólise mais difícil de ocorrer), mas o mais conveniente é tratá-la com hidrazina em etanol em refluxo, resultando em uma amina primária.

A partir de aminação redutiva
As aminas podem ser sintetizadas em uma única etapa através da aminação redutiva, que se dá pelo tratamento de aldeído ou cetona com amônia ou amina na presença de um agente redutor.

O mecanismo de reação da aminação redutiva ocorre de acordo com a Figura 52. A amônia, através de uma reação de adição nucleofílica, se une ao grupo carbonila da cetona, formando um intermediário carbinolamina. Este intermediário perderá uma molécula de água, dando origem a uma imina que será reduzida a uma amina utilizando NaBH4 ou H2/Ni.

Os agentes redutores que podem ser utilizados, além dos já citados, são NaBH3CN ou LiBH3CN, similares ao NaBH4 (Figura 53).

A partir de redução de nitrilas, amidas e nitrocompostos
A formação de uma amina primária pode ocorrer através da redução de uma nitrila com LiAlH4, onde a adição nucleofílica do íon hidreto à ligação polar CΞN forma um ânion imina. A imina formada ainda possui uma ligação C=N e, assim, sofre a segunda adição nucleofílica de hidreto para formar um diânion.
Esta segunda adição só é possível porque o intermediário monoânion é estabilizado por complexação ácido-base de Lewis a uma espécie de alumínio (AlCl3, por exemplo). Em seguida, ocorre a protonação do diânion pela adição de água, gerando uma amina.

Já a redução de amidas com LiAlH4 é um excelente método de conversão de ácidos carboxílicos e seus derivados em aminas primárias, secundárias ou terciárias. A redução da amida ocorre pela adição nucleofílica de íon hidreto ao grupo carbonila da amida, seguida da liberação do átomo de oxigênio como um grupo abandonador ânion aluminato, formando o íon imínio intermediário que sofrerá redução para a formação da amina.

Para o preparo de aminas aromáticas, o método mais utilizado é a nitração do anel aromático, aplicável a uma variedade de compostos aromáticos, seguida da redução do grupo nitro. Os métodos mais frequentes para redução do grupo nitro são a hidrogenação catalítica ou o tratamento do grupo nitro com ácido e ferro, zinco, estanho ou cloreto de estanho.
Hidrogenação catalítica
A hidrogenação catalítica, geralmente, é incompatível com a presença de qualquer outro grupo que possa sofrer redução (por exemplo, C=C ou carbonila).
Cloreto de estanho II
Já o cloreto de estanho II (SnCl2) é geralmente utilizado quando há outros grupos funcionais passíveis de redução.


Rearranjos de hofmann e curtius
O rearranjo de Hofmann ocorre quando uma amida primária é tratada com solução de bromo ou cloro e base, gerando aminas. O mecanismo para esta reação é mostrado na Figura 57.

Etapa 01
Inicialmente, a amida sofre a bromação promovida por base, o ânion formado irá reagir com o bromo através de uma reação de substituição α, formando a bromoamida.
Etapa 02
Em seguida, ocorre a remoção do próton remanescente por uma base levando à formação do ânion bromoamida.
Etapa 03
O ânion bromoamida sofre rearranjo quando o grupo R ligado ao átomo de carbono da carbonila migra para o átomo de nitrogênio ao mesmo tempo em que o íon brometo deixa a molécula, produzido um isocianato.
Etapa 04
O isocianato é rapidamente hidrolisado pela base aquosa em um íon carbamato que sofre descarboxilação espontânea resultando na formação da amina.
Um ponto importante a ser observado é que nas duas primeiras etapas do mecanismo, dois átomos de hidrogênio devem estar presentes no nitrogênio da amida para que a reação possa ocorrer. Logo, o rearranjo de Hofmann é limitado ao uso de amidas primárias.
O rearranjo de Curtius, embora envolva uma azida de acila, possui mecanismo semelhante ao rearranjo de Hofmann. No rearranjo de Curtius, o grupo R migra do carbono acila para o átomo de nitrogênio à medida que o grupo abandonador sai. A reação ocorre com aquecimento de uma azida de acila, preparada através de uma ração de substituição nucleofílica de acila de um cloreto de ácido com azida de sódio.

A seguir, temos dois exemplos de compostos utilizados comercialmente sintetizados a partir do rearranjo de Hofmann e Curtius.
O primeiro é o supressor de apetite fentermina, preparado por rearranjo de Hofmann de uma amida primária.
O segundo é o antidepressivo tranilcipromina, preparado por rearranjo de Curtius do cloreto de 2-feniliciclopropanocarbonila.

REAÇÕES DE AMINA
As aminas são muito importantes em diversos tipos de reação, podendo agir como base, como nucleófilos nas reações de alquilação e nas reações de acilação e como grupo ativador em anéis aromáticos orientando nas posições orto e para.
Acilação
As aminas primárias e secundárias podem ser transformadas em amidas através de uma reação de substituição de acila com anidrido acético ou cloreto de ácido. Neste caso, a superacilação do nitrogênio não ocorre, pois o produto de amida é muito menos nucleofílico e menos reativo que a amina de partida.

Eliminação de hofmann
As aminas podem ser convertidas em alcenos através de uma reação de eliminação, entretanto, íon amideto não é um bom grupo abandonador, sendo necessária sua conversão em um melhor grupo abandonador. Esta estratégia é utilizada na Eliminação de Hofmann, onde a amina é metilada para produzir um haleto de amônio quaternário que irá sofrer a reação de eliminação para formar o alceno sob aquecimento com uma base.

A base utilizada, geralmente, é o óxido de prata que atua trocando o íon hidróxido por iodeto no hidróxido de amônio quaternário, fornecendo a base necessária para provocar a eliminação. No caso das eliminações de Hofmann que envolvem substratos carregados, o produto formado seguirá a regra de Hofmann, ou seja, o alceno formado será o menos substituído.
A razão dessa seletividade, provavelmente, é estérica, ou seja, em função do tamanho do grupo abandonador, triaquilamina, a base deve retirar o hidrogênio da posição mais livre estericamente, ou seja, menos impedida (MCMURRY, 2011).
Substituição aromática eletrofílica
Como mencionado anteriormente, o grupo amino é fortemente ativador e orientador orto e para em reações de substituição aromática eletrofílica, entretanto essa alta reatividade pode ocasionar uma polissubstituição. Outro ponto negativo é que as reações de Friedel-Crafts em benzenos amino-substituídos não são bem-sucedidas, pois o grupo amino forma um complexo ácido-base com o catalisador AlCl3, que impede o andamento da reação.
Esses fatores, a alta reatividade e a basicidade da amina podem ser resolvidos realizando as reações de substituição aromática eletrofílica na amida correspondente ao invés da amina livre. Os substituintes amido, embora seja ativador e orientador orto e para, são mais fracos e menos básicos que o grupo amino, pois o par de elétron na amida está mais deslocalizado.
Inicialmente, a amina é tratada com anidrido acético formando o amideto de acetila correspondente, ou acetamida.
Em seguida, ocorre a bromação, dando origem a um único produto que será hidrolisado com solução aquosa básica, produzindo a amina livre. As reações de alquilação e acilação de Friedel-Crafts também ocorrem normalmente, convertendo a amina em amidas.

Reações de aminas com ácido nitroso
O ácido nitroso é capaz de reagir com todas as classes de aminas, entretanto o resultado obtido dependerá se a amina é primária, secundária ou terciária.
Nas reações de diazotação, as aminas alifáticas primárias reagem com o ácido nitroso, formando sais de diazônio alifáticos. Esses sais, mesmo a temperaturas baixas, são altamente instáveis e decompõem-se espontaneamente através da perda de nitrogênio para formar carbocátions.
Atenção
Entretanto, essas reações possuem pequena importância sintética, uma vez que produzem uma mistura complexa de produtos, a partir do carbocátion podemos formar alcenos, álcoois e haletos de alquila através da remoção de um próton, reação com água e reação com X-.

Já as aminas secundárias (arila e alquila) reagem com o ácido nitroso para produzir as N-nitrosaminas, enquanto as aminas alifáticas terciárias formam o sal de amina e um composto N-nitrosoamônio.
As arilaminas terciárias reagem com ácido nitroso para formar compostos aromáticos C-nitroso.
A nitrosação irá ocorrer quase exclusivamente na posição para ou na posição orto, caso a posição para não esteja livre.

Mas e a reação com arilaminas primárias? Essa, de longe, é a mais importante das reações de aminas com ácido nitroso, por isso, iremos falar apenas sobre elas no item Reações de substituição de sais de arenodiazônio.
N-Nitrosaminas
As N-nitrosaminas são compostos comumente encontrados na água, em alimentos defumados e grelhados, laticínios e vegetais. Entretanto, a exposição desses compostos acima dos limites aceitáveis e por longo período pode ocasionar agravos à saúde, aumentando o risco de ocorrência de câncer.
Saiba mais
Em 2018, o tema nitrosamina ficou em evidência na indústria farmacêutica e na Agência Nacional de Vigilância Sanitária (Anvisa), pois níveis acima do critério de aceitação de nitrosaminas foram encontrados em alguns medicamentos para pressão arterial, conhecidos como “sartanas”, ocasionando o recolhimento de vários produtos em vários países.
A partir daí, Anvisa iniciou uma investigação no Brasil e detectou níveis inaceitáveis de nitrosaminas nos medicamentos, determinando o recolhimento dos lotes de medicamentos, proibição de importação de alguns insumos farmacêuticos ativos e a publicação da RDC 283/2019 – que estabelece regras para investigação, controle e eliminação de nitrosaminas (Anvisa, nota informativa 1/2020).
Segundo essa nota da Anvisa e o guia do Food and Drug Administration (FDA, 2020), a presença das impurezas no medicamento pode ser oriunda da presença simultânea de nitritos e aminas na síntese dos insumos farmacêuticos, materiais de partida, reagentes ou solventes.
O FDA (2020) identificou sete impurezas de nitrosamina que teoricamente poderiam estar presentes em medicamentos: N-nitrosodimetilamina (NDMA), N-nitrosodietilamina (NDEA), ácido N-nitroso-N-metil-4-aminobutírico (NMBA), N-nitrosoetilisopropilamina (EIPNA), N- nitrosodiisopropilamina (NDIPA), N-nitrosodibutilamina (NDBA) e N-nitrosometilfenilamina (NMPA) (Figura 65). Cinco deles (NDMA, NDEA, NMBA, EIPNA e NMPA) foram realmente detectados em insumos ou medicamentos.

Reações de substituição de sais de arenodiazônio
As arilaminas primárias reagem com ácido nitroso para formar sais de arenodiazônio, entretanto muitos desses sais são instáveis à temperatura acima de 5-10°C. Mas, felizmente, em muitas reações de substituição de sais de diazônio, não há necessidade de que eles sejam isolados, podendo ser adicionados outros reagentes no meio reacional, com exceção da substituição do grupo diazônio pelo –F (neste caso é preciso isolar o sal).

Neste tipo de reação, os sais de arenodiazônio são extremamente úteis porque o grupo diazônio pode ser substituído por um nucleófilo em uma reação de substituição, podendo reagir com o cloreto cuproso, brometo cuproso, e cianeto cuproso para fornecer os respectivos produtos nos quais o grupo diazônio foi substituído por –Cl, -Br e –CN. Os mecanismos dessas reações não são completamente entendidos, mas parecem ser radicalares por natureza, não iônicas.
A seguir, teremos exemplos de reações de substituição de sais de diazônio utilizando diferentes reagentes. No primeiro exemplo, podemos reagir o sal de diazônio com o iodeto de potássio para fornecer produtos nos quais o grupo diazônio foi substituído pelo iodo.

No segundo exemplo, o sal de diazônio é substituído pelo flúor através do tratamento com ácido fluorobórico (HBF4). Neste caso, o fluorato de diazônio é isolado, seco e aquecido até a decomposição e formação do fluoreto de arila.

Por fim, temos a substituição do sal de diazônio pela hidroxila através da adição de óxido cuproso a solução diluída do sal de diazônio com excesso de nitrato cúprico.

Reações de acoplamento de sais de arenodiazônio
As reações de acoplamento dos sais de arenodiazônio são substituições eletrofílicas típicas nas quais os íons diazônios, eletrófilos fracos, reagem com compostos aromáticos altamente reativos, fenóis e arilaminas terciárias, formando compostos azo, produtos coloridos e de muito brilho.

O acoplamento entre os cátions de arenodiazônio e os fenóis ocorrem facilmente em soluções ligeiramente alcalina (pH 7-10), para que haja uma quantidade relevante do íon fenóxido (mais reativos que os fenóis). Entretanto, se a solução estiver com pH>10, o próprio sal de arenodiazônio reage com o íon hidróxido para formar um diazoidróxido ou íon diazotato. Enquanto com as aminas a reação ocorre mais facilmente em soluções ligeiramente ácidas (pH 5-7), pois, nessas condições, a concentração do cátion está no máximo e uma quantidade excessiva de amina não foi convertida em um sal não reativo. O acoplamento ocorre quase que exclusivamente na posição para, se estiver disponível, ou na posição orto, se a posição para não estiver disponível.

Síntese de Sulfonamidas e sua importância na Indústria Farmacêutica
Verificando o aprendizado
ATENÇÃO!
Para desbloquear o próximo módulo, é necessário que você responda corretamente a uma das seguintes questões:
O conteúdo ainda não acabou.
Clique aqui e retorne para saber como desbloquear.
Conclusão
Considerações Finais
Neste tema, abordamos as principais reações envolvendo os compostos oxigenados não carbonilados e aminas, e discutimos os princípios mecanísticos de cada um. Por fim, apresentamos as principais fontes e uso destes compostos.
Ao concluir o estudo deste tema, você deve ter percebido que os compostos oxigenados não carbonilados e aminas possuem usos bastantes diversificados e estão presentes no nosso cotidiano, seja através de fármacos sintéticos ou da natureza.