Descrição
Características químicas e físicas dos ácidos carboxílicos e derivados e suas aplicações.
PROPÓSITO
Obter conhecimento e informações básicas sobre estrutura, nomenclatura, propriedades, métodos de obtenção e reações de ácidos carboxílicos, bem como de seus derivados, com ênfase nos mecanismos das reações, é importante para prever as características e a obtenção de compostos orgânicos dessa classe com aplicações nos setores químico e farmacêutico.
OBJETIVOS
Módulo 1
Descrever as características químicas e físicas, reatividade e acidez dos ácidos carboxílicos e derivados
Módulo 2
Descrever os princípios mecanísticos gerais e as principais reações de substituição nucleofílica acílica
Módulo 3
Reconhecer as reações que envolvam a substituição do hidrogênio alfa (Hα) por eletrófilos e os princípios mecanísticos gerais da condensação de Claisen e da descarboxilação aplicada ao ácido malônico
Introdução
Os ácidos carboxílicos e seus derivados estão entre os mais comuns de todas as moléculas, tanto no laboratório quanto nas rotas biológicas, sendo comumente encontrados na estrutura de diversas biomoléculas essenciais à vida. Além disso, estão presentes no nosso cotidiano quando se trata das indústrias farmacêutica, química e de alimentos.
Neste tema, você terá a oportunidade de reconhecer as principais reações que envolvem os ácidos carboxílicos e derivados, assim como de descrever os princípios mecanísticos de cada tipo de reação. Abordaremos também alguns exemplos de reações e suas aplicações no nosso cotidiano.
MÓDULO 1
Descrever as características químicas e físicas, reatividade e acidez dos ácidos carboxílicos e derivados
Introdução à química dos ácidos carboxílicos
Os ácidos carboxílicos ocupam um lugar central entre os compostos de carbonila, sendo comumente encontrados na química e na bioquímica. Além disso, são materiais de partida na síntese de derivados de acila, como cloretos ácidos, ésteres, amidas e tioésteres.
No universo bioquímico podemos citar os aminoácidos presentes em nossa dieta alimentar, tais como o ácido L-glutâmico e o ácido γ-aminobutírico (GABA), que atuam como neurotransmissores importantes no sistema nervoso. Podemos citar, também, as prostaglandinas, que exercem papéis importantes em muitos processos fisiológicos, como, por exemplo, o controle da pressão sanguínea, o processo inflamatório e a atividade do sistema digestivo.
COSTA et al., 2003.
Na imagem a seguir, você pode ver a representação de fórmulas químicas das prostaglandinas dos tipos D, E e F.

Na natureza podemos encontrá-los nas plantas e nas frutas. O ácido oxálico, por exemplo, é um ácido dicarboxílico tóxico que pode estar presente no espinafre. Mas não se assuste! A sua concentração no espinafre é muito baixa para representar qualquer perigo; além disso, o ácido oxálico é um ótimo removedor de manchas e ferrugens. Nas frutas, podemos encontrar os ácidos cítrico, málico e tartárico, cujos sabores são acentuados e estão presentes em limões, maçãs e uvas, respectivamente.
Como explicado por COSTA et al., 2003, na indústria alimentícia, utilizamos, por exemplo, o glutamato monossódico – para acentuar o sabor de uma série de produtos –, o ácido benzóico – como preservativo de alimentos – e o ácido acético, presente no vinagre – usado na alimentação e no preparo de ketchup, picles e molhos.
Como podemos ver, a compreensão das propriedades e reações dos ácidos carboxílicos é fundamental para entendermos a Química Orgânica, uma vez que eles estão presentes em muitos processos industriais e classes de produtos bioativos, além de serem utilizados como intermediários na síntese de outros compostos.
Estrutura e propriedades dos ácidos carboxílicos
Na química orgânica, ácidos carboxílicos têm um grupo funcional formado pela presença de uma carbonila e de uma hidroxila (-COOH), denominado grupo carboxila (figura 1).

Na estrutura do grupo carboxila, o carbono apresenta hibridização sp2, em que os três átomos ligados ao átomo de carbono ocupam o mesmo plano, com ângulos de ligação C-C=O e O=C-O de aproximadamente 120° (tabela 1). Possuem dois comprimentos de ligação carbono-oxigênio, sendo o comprimento de ligação C=O mais curto, quando comparado à ligação C-O. (MCMURRY, 2011)
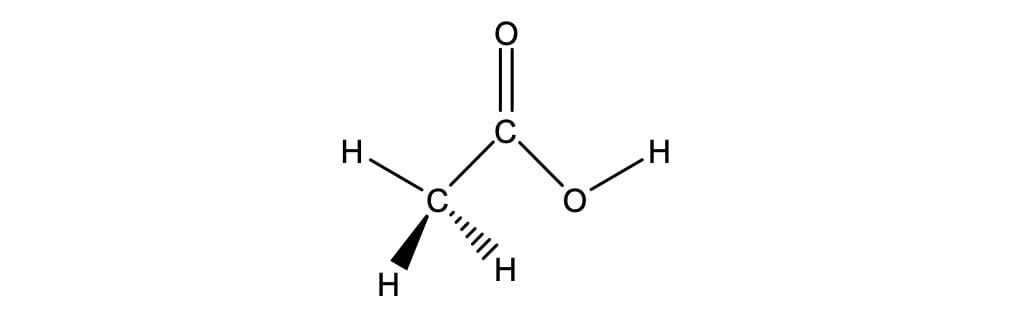
|
|||
| Ângulos de ligação | Graus | Comprimento de ligação | pm |
|---|---|---|---|
| C-C=O | 119 | C-C | 152 |
| C-C-OH | 119 | C=O | 125 |
| O=C-OH | 122 | C-OH | 131 |
Os ácidos carboxílicos podem reagir com bases, perdendo um próton para formar sais de carboxilato. Essa reação ocorre pela quebra heterolítica da ligação O-H, ficando os elétrons no átomo de oxigênio (figura 2).

Esses compostos são substâncias polares, podendo formar ligações de hidrogênio intermoleculares intensas (formação de dímeros cíclicos), proporcionando elevados pontos de ebulição. Já a solubilidade em água decorre da formação de ligações de hidrogênio com a água, fazendo com que compostos com baixa massa molecular apresentem notável miscibilidade com a água (tabela 2).
| Nome IUPAC | Nome comum | Estrutura | p.f. (°C) | p.e. (°C) | Solubilidade em água* | pKa |
|---|---|---|---|---|---|---|
| Saturados | ||||||
| Ácido metanoico | Ácido fórmico | HCO2H | 8 | 101 | ∞ | 3,75 |
| Ácido etanoico | Ácido acético | CH3CO2H | 17 | 118 | ∞ | 4,76 |
| Ácido butanoico | Ácido butírico | CH3CH2CH2CO2H | -6 | 163 | ∞ | 4,81 |
| Ácido 3-metilbutanoico | Ácido isovalérico | (CH3)2CHCH2COOH | -29 | 177 | 5,0 | - |
| Ácido hexanoico | Ácido caproico | CH3(CH2)4CO2H | -3 | 205 | 1,08 | 4,84 |
| Ácido decanoico | Ácido cáprico | CH3(CH2)8CO2H | 31 | 269 | 0,015 | 4,84 |
| Insaturados | ||||||
| Ácido 2-propenoico | Ácido acrílico | CH2=CHCO2H | 14 | 141 | 8 | - |
| Ácido benzoico | Ácido benzoico | C6H5CO2H | 122 | 250 | 0,34 | 4,19 |
| Ácidos graxos | ||||||
| Ácido dodecanoico | Ácido láurico | CH3(CH2)10CO2H | 44 | 179 | 0,006 | 5,30 |
| Ácido tetradecanoico | Ácido mirístico | CH3(CH2)12CO2H | 59 | 200 | 0,002 | - |
| Ácido hexadecanoico | Ácido palmítico | CH3(CH2)14CO2H | 63 | 219 | 0,0007 | 6,46 |
| Ácido octadecanoico | Ácido esteárico | CH3(CH2)16CO2H | 70 | 383 | 0,0003 | - |
| Ácidos dicarboxílicos | ||||||
| Ácido etanodioico | Ácido oxálico | HO2C-CO2H | 189 | - | 9,0 | - |
| Ácido propanodioico | Ácido malônico | HO2C-CH2-CO2H | 136 | - | 74 | - |
| Ácido cis-2-butenodioico | Ácido maleico | Cis-HO2CCH=CHCO2H | 140 | - | 79 | - |
| *g 100 mL-1 de água, 25°C | ||||||
De acordo com a tabela 2, é possível observar que ácidos carboxílicos de baixo peso molecular são líquidos e miscíveis com água devido à solvatação pela formação de ligações de hidrogênio com o solvente; já seus homólogos superiores são sólidos em temperatura ambiente e insolúveis em água, pois na medida em que aumenta o tamanho da cadeia hidrocarbônica, a solubilidade em água diminui.
Dentre os homólogos superiores, estão os ácidos graxos (C12-C20), amplamente encontrados na natureza sob a forma de ésteres do glicerol, sendo conhecidos como triglicerídeos. Os ácidos graxos podem apresentar cadeias saturadas, sólidos em temperatura ambiente, ou insaturadas de configuração cis, em geral óleos e gorduras, como os ácidos oleico, linoleico e linolênico.
Na figura 3, é possível observar que, nos ácidos graxos insaturados, a presença de uma ligação dupla cis impede a formação de uma rede cristalina estável, proporcionado os baixos pontos de fusão desses compostos.

IUPAC
União Internacional de Química Pura e Aplicada que estabelece as regras de nomenclatura de compostos orgânicos.
Quebra heterolítica
Cisão/Quebra heterolítica: é a ruptura eletronicamente desigual da ligação entre dois átomos, onde apenas um deles (geralmente o mais eletronegativo) fica com o par de elétrons.
Dímeros cíclicos
Que é composto de duas partes.
Molécula resultante da combinação de duas moléculas idênticas.
Nomenclatura dos ácidos carboxílicos
No sistema IUPAC, os ácidos carboxílicos derivados de hidrocarbonetos de cadeia aberta são sistematicamente nomeados substituindo-se o sufixo -o da maior cadeia hidrocarbônica que contém a carboxila pelo sufixo -oico, precedido da palavra ácido. Apesar dos nomes sistemáticos ou substitutivos IUPAC, muitos ácidos carboxílicos apresentam nomes comuns derivados do latim ou grego que indicam uma das suas fontes naturais (figura 4).

Para nomear compostos com cadeias ramificadas e/ou com ligações múltiplas (dupla ou tripla), devemos numerar a cadeia principal, e o carbono do grupo -COOH sempre será a posição 1, veja a seguir na figura 5.

Quando o grupo -COOH estiver ligado a um anel, ele será considerado um substituinte da cadeia principal e, nesses casos, o carbono do grupo -COOH será ligado ao C1 e não será numerado nesse sistema. Por exemplo, um ciclobutano contendo COOH como substituinte é chamado de ácido ciclobutanocarboxílico. Já os ácidos carboxílicos aromáticos são nomeados como derivados do anel aromático correspondente e pode-se utilizar prefixos orto- (o-), meta- (m-) e para- (p-) para designar a posição dos substituintes. Veja alguns exemplos na figura 6.

Vamos ver uma curiosidade sobre o ácido benzóico?
O ácido benzóico faz parte da composição natural de diversos alimentos, como:

Cogumelos

Tomate

Iogurte

Cerveja

Vinho

Maça

Uva
Outra opção de nomenclatura para designar as posições dos substituintes na cadeia principal é a utilização das letras gregas alfa (α), beta (β), gama (γ), delta (δ), épsilon (ε) e ômega (ω).
A letra α (alfa) é a primeira a ser utilizada no carbono adjacente à carboxila, seguida pelas demais, β, γ, δ e ε. Já a posição ω será sempre o carbono mais afastado da carboxila, independentemente do número de carbonos da cadeia principal.
Já os ácidos dicarboxílicos, também chamados de diácidos, apresentam dois grupos –COOH. Para os ácidos alifáticos, a nomenclatura deve ser realizada pela adição do sufixo –dioico ao nome do alcano da cadeia principal (cadeia que possui os dois grupos –COOH), precedido pela palavra ácido. A cadeia principal será numerada a partir do grupo –COOH que permitir a menor numeração para os substituintes. Para os ácidos cíclicos, consideram-se as carboxilas substituintes da estrutura cíclica.

Reatividade química e acidez dos ácidos carboxílicos e seus derivados
Acidez do grupo carboxílico
Como o próprio nome diz, a propriedade principal dos ácidos carboxílicos é a acidez. Os ácidos carboxílicos são ácidos de Brønsted-Lowry que se dissociam levemente em água para gerar o hidrônio, H3O+, e os ânions carboxilatos, RCO2-.
Para a maioria dos ácidos carboxílicos, a constante de acidez, Ka é aproximadamente 10-4 a 10-5, mostrando serem ácidos muitos mais fracos do que os ácidos inorgânicos, porém, muito mais fortes que os álcoois. Temos, como exemplo, o etanol, que possui Ka de aproximadamente 10-16, tornando-o mais fraco do que o ácido acético que possui Ka igual a 1,34 x 10-5.
Constante de acidez (Ka)
A constante de acidez (Ka) é proporcional à concentração dos íons formados. Portanto, quanto maior o valor de Ka mais ionizado é o ácido; consequentemente, maior a sua força. Logo, quanto mais forte o ácido, maior o valor de Ka, menor o valor de pKa e mais fraca a sua base conjugada.
Segundo a teoria Brønsted-Lowry, ácidos são espécies (íons ou moléculas) doadoras de prótons (H+) e bases são espécies aceptoras de prótons. Um ácido reage com uma base, levando à formação de um ácido conjugado da base e uma base conjugada do ácido.
Em ambos os casos, a acidez é decorrente do fato de a base conjugada do ácido ser estabilizada por ter sua carga negativa em um átomo de oxigênio fortemente eletronegativo. Entretanto, o etanol dissocia-se, produzindo um íon etóxido, no qual a carga negativa está localizada em um átomo eletronegativo, enquanto o ácido acético gera um íon acetato, no qual a carga negativa está deslocalizada sobre dois átomos de oxigênio equivalentes. Sendo assim, o íon acetato é mais estável do que o íon etóxido, devido ao efeito de ressonância.
O íon acetato apresenta duas estruturas canônias de mesma energia, distribuindo a carga negativa entre dois átomos eletronegativos, enquanto no etóxido um único átomo de oxigênio porta a carga negativa (figura 8). O efeito de ressonância torna o íon acetato mais estável que o íon etóxido, possuindo uma energia mais baixa e sendo mais favorecido no equilíbrio de dissociação.

Mas, por que o íon fenolato é menos estável do que o íon acetato, uma vez que o íon fenolato apresenta mais estruturas de ressonância?
A resposta fica clara se observarmos a figura 9.
No fenolato, três das formas canônicas mostram a carga negativa situada em átomos de carbono, que apresentam menor contribuição, ou seja, são menos eletronegativos e suportam menos a carga negativa.

Efeito do substituinte sobre a acidez
Como a dissociação de um ácido carboxílico é um processo de equilíbrio, qualquer fator que estabilize o ânion carboxilato irá favorecer o deslocamento do equilíbrio no sentido da dissociação, aumentando a acidez. Utilizaremos como base, para discussão deste tópico, os compostos da figura 10, a seguir.

Observe na figura que os compostos que têm o cloro como substituinte apresentam um menor valor de pKa em relação aos seus correspondentes sem o substituinte cloro. No ácido α-clorobutírico, por exemplo, o caráter eletronegativo do cloro é o responsável por “puxar” elétrons da ligação C-Cl. O carbono, por sua vez, puxa elétrons do carbono da carboxila, que irá puxar elétrons dos átomos de oxigênio, estabilizando o carboxilato. Sendo assim, quanto maior o número de átomos de cloro em um mesmo carbono, menor será o pka do composto e, consequentemente, maior a acidez.
Entretanto, observe: à medida que o átomo de cloro se afasta da posição α, os valores de pKa aumentam, pois o efeito indutivo é transmitido por meio das ligações σ adicionais, diminuindo a sua intensidade.
Como pôde ser visto na figura 9, o ácido α-fluoracético é o mais ácido entre os ácidos α-substituídos por halogênios. Esse fato é decorrente do maior efeito polar do flúor sobre a estabilização do carboxilato, ou seja, quanto maior a eletronegatividade do substituinte, maior a acidez do composto.
Os efeitos dos substituintes na acidez também são encontrados nos ácidos benzoicos substituídos. Quando os sistemas aromáticos são substituídos por grupos doadores de elétrons (CH3, OH, CH3O, NH2, N(CH3)2 ), tanto a reatividade quanto a acidez irão diminuir em relação ao ácido benzoico sem substituintes. Por outro lado, substituições por grupos retiradores de elétrons (CF3, NO2, CN, CHO, Cl) irão aumentar a eletrofilicidade da carbonila e sua acidez em relação ao produto não substituído.

Reatividade química dos ácidos carboxílicos e derivados
Os fatores estéricos e eletrônicos são de extrema importância na determinação da reatividade dos ácidos carboxílicos e seus derivados.
Na figura 11, podemos observar que, ao compararmos uma série de ácidos similares, os compostos com menor número de substituintes são mais reativos. Essa reatividade ocorre porque os grupos carbonila estão mais desimpedidos estericamente.

Do ponto de vista eletrônico, a reatividade química frente a nucleófilos depende da contribuição relativa dessas estruturas, as quais dependem da natureza do grupo substituinte X. Por exemplo, os cloretos de ácidos são mais reativos do que as amidas e os ésteres, pois o cloro, por ser mais eletronegativo, terá mais força para retirar elétrons do carbono da carbonila, tornando-a mais δ+. Além disso, nesse caso, dentre as suas formas canônicas, a contribuição da forma canônica C – onde todos os átomos estão com o octeto completo e o carbono encontra-se neutro, o que diminui a eletrofilicidade – será muito pequena (figura 12).

Quando se trata de ésteres e amidas, a contribuição da estrutura C é mais expressiva em ambos os compostos. Entretanto, a menor eletronegatividade do nitrogênio em relação ao oxigênio faz com que o nitrogênio suporte melhor a carga positiva, aumentando a contribuição da estrutura C; sendo assim, os ésteres serão mais reativos do que as amidas.
Agora, ao compararmos os tioésteres com os ésteres correspondentes, veremos que os primeiros são mais reativos, pois a interação por ressonância dos pares de elétrons não compartilhados do enxofre com a ligação C=O é pouco efetiva. Sendo assim, a contribuição da forma canônica C nos tioésteres será pequena, prevalecendo a estrutura B, na qual o carbono está positivo e altamente eletrofílico (figura 13).

Como consequência dessas diferenças de reatividade, podemos dizer que um derivado ácido mais reativo pode ser convertido diretamente em um menos reativo, mas o inverso geralmente é mais difícil e, quando possível, requer reagentes especiais.
Assista agora o vídeo sobre Nomenclatura e reatividades dos ácidos carboxílicos e derivados.
Verificando o aprendizado
ATENÇÃO!
Para desbloquear o próximo módulo, é necessário que você responda corretamente a uma das seguintes questões:
O conteúdo ainda não acabou.
Clique aqui e retorne para saber como desbloquear.
MÓDULO 2
Descrever os princípios mecanísticos gerais e as principais reações de substituição nucleofílica acílica
Reação de substituição nucleofílica acílica
A substituição nucleofílica de acila descreve uma classe de reações de substituição envolvendo nucleófilos, espécie química que doa um par de elétrons e compostos derivados de ácidos carboxílicos.
Nesse tipo de reação, os derivados de ácidos carboxílicos apresentam um carbono da acila ligado ao grupo abandonador – X, que pode sair como um ânion estável. Sendo assim, quando um nucleófilo é adicionado ao composto de acila, o intermediário tetraédrico é formado e o grupo abandonador é eliminado, gerando um novo composto de acila (figura 1).

Embora a reação total da substituição nucleofílica de acila seja superficialmente similar ao tipo de substituição nucleofílica que ocorre durante uma reação SN2, os mecanismos das duas reações são bem diferentes. A reação SN2 ocorre em uma única etapa e o nucleófilo ataca por trás, ou seja, pelo lado diretamente oposto ao grupo abandonador; já na substituição nucleofílica de acila, a reação ocorre em duas etapas, envolvendo a formação de um intermediário tetraédrico.
Reação SN2
Reação de substituição nucleofílica bimolecular.
O processo como um todo, no caso das substituições de acila, ocorre por intermédio de um mecanismo de adição nucleofílica-eliminação, podendo acontecer em meio neutro, ácido ou básico. Entretanto, os meios básico e ácido são as condições mais comuns, pois aceleram significativamente a velocidade da reação (figura 2).

Na figura 2, temos um exemplo de uma reação de substituição nucleofílica de acila a partir de um cloreto de ácido.
Abaixo veremos as etapas dessa reação:

Adição do nucleófilo ao grupo carbonila eletrofílico

Formação do intermediário tetraédrico instável

Reação de eliminação do íon cloreto para formação do éster
Saiba mais
A presença da base, como, por exemplo, a piridina, será importante para a remoção do próton do álcool adicionado à carbonila.
Podemos observar, também, que as substituições em grupos carbonil trigonais passam por um intermediário tetraédrico e depois para um produto trigonal.
Chamamos o intermediário de tetraédrico pois o átomo de carbono trigonal – configuração sp2 – da carbonila passa a ser um carbono tetraédrico – configuração sp3.
Conversão de ácidos carboxílicos em cloretos de ácido
No módulo anterior, vimos que, devido às diferenças de reatividade, para que um derivado ácido mais reativo seja convertido em um menos reativo, é necessária a utilização de reagentes especiais que transformem um grupo abandonador ruim em um grupo abandonador melhor do que aquele presente no produto da reação.
Os ácidos carboxílicos podem ser convertidos em cloretos de ácido pelo tratamento com o cloreto de tionila (SOCl2), tricloreto de fósforo (PCl3) ou pentacloreto de fósforo (PCl5), em ambos os casos as reações envolvem adição nucleofílica-eliminação de um íon cloreto em um intermediário altamente reativo: um clorossulfito de acila protonado, um clorofosfito de acila protonado ou um clorofosfato de acila protonado (Figura 3).

Como podemos observar no mecanismo de reação, o ácido carboxílico irá atacar o enxofre do cloreto de tionila por este ser eletrofílico, uma vez que apresenta um átomo de oxigênio e dois cloros ligados a ele, formando um intermediário instável e altamente eletrofílico. A reprotonação do intermediário instável irá proporcionar-lhe maior eletrofilicidade para que ocorra a reação, mesmo com um nucleófilo fraco Cl-.
Reações de cloretos de ácidos
Uma vez preparado, o cloreto de ácido ou cloreto de acila, o mais reativo dentre os derivados de ácidos carboxílicos, reage facilmente e com bons rendimentos com vários nucleófilos, podendo ser convertido aos derivados menos reativos.
Sendo assim, devido a sua reatividade, na maioria dos casos a melhor rota sintética para um anidrido, amida ou éster envolverá a síntese inicial do cloreto de acila do ácido e, posteriormente, a conversão ao derivado ácido desejado (figura 4).

Além das reações representadas na figura 4, os cloretos de acila podem reagir com água ou com base aquosa – reação denominada hidrólise –, mas essas reações normalmente são pouco realizadas, pois regeneram o ácido carboxílico, destruindo a utilidade do reagente cloreto de acila. Nesse caso, a utilização da base tem como objetivo remover o ácido clorídrico formado e evitar que ele provoque reações laterais (figura 5).

A reação de cloretos de ácidos com álcoois, denominada alcoólise, provavelmente é o método mais comum para preparação de ésteres em laboratório. As reações são geralmente realizadas na presença de piridina ou hidróxido de sódio (NaOH) para reagir com o ácido formado, assim como ocorre na hidrólise.
Nesse caso, a escolha do álcool a ser utilizado irá influenciar na velocidade da reação, pois álcoois com grupos mais volumosos tornam a reação consideravelmente mais lenta, devido ao impedimento estérico, resultando em uma ordem de reatividade: álcool primário > álcool secundário > álcool terciário. Na figura 6, podemos observar que é possível esterificar um álcool na presença de um álcool mais impedido. Nesse caso, o álcool secundário é mais impedido e menos reativo, enquanto o álcool primário é menos impedido e mais reativo.

Para a síntese de amidas, podemos reagir o cloreto de ácido com amônia ou aminas; entretanto, apenas aminas mono- e dissubstituídas podem ser utilizadas. Assim como nas reações anteriores, para a síntese de amidas ocorre a formação de HCl, sendo necessário neutralizá-lo. Para isso, são utilizados dois equivalentes de aminas: um para reagir com o cloreto de ácido e outro para neutralizar o HCl, formando o sal de cloreto de amônio. Contudo, dependo do valor agregado da amina, pode-se utilizar um equivalente de uma base barata, como, por exemplo, o hidróxido de sódio.
Conversão de ácidos carboxílicos em ésteres
Reações de esterificações de Fischer são caracterizadas pela síntese de ésteres a partir de ácidos carboxílicos e álcoois. Elas ocorrem muito lentamente, pois os ácidos carboxílicos não são reativos o suficiente para sofrerem diretamente uma adição nucleofílica, porém atingem o equilíbrio em poucas horas quando uma pequena quantidade de ácido forte, HCl ou H2SO4, é adicionada ao meio.
O ácido inorgânico é responsável por protonar uma pequena porcentagem de átomo de oxigênio do grupo carbonila, tornando-a extremamente suscetível ao ataque, mesmo por um nucleófilo fraco como o álcool. O álcool realiza o ataque nucleofílico, formando o intermediário tetraédrico instável. Note que nenhum dos grupos de saída (R, HO, RO) do intermediário é muito bom; sendo assim, a ajuda está novamente no ácido que irá protonar o oxigênio (OH), convertendo-o em um bom grupo de saída (água). A perda de um próton e a expulsão de H2O regeneram o catalisador ácido e geram o éster como produto.
O mecanismo da reação é mostrado na Figura 7.

Uma vez que a posição de equilíbrio controla a quantidade de éster formada, ela será favorecida quando um excesso de álcool for utilizado como solvente. Entretanto, a formação de um ácido carboxílico será beneficiada quando um excesso de água estiver presente. Você poderá ver na figura 8.
Assim, o resultado a ser obtido dessa reação irá depender das condições escolhidas.
Utilize excesso de álcool e, se possível, remova a água à medida que ela seja formada.
Utilize de um grande excesso de água (refluxo com uma solução aquosa de HCl ou H2SO4).

Observe, no mecanismo de reação, que:
A hidrólise de éster catalisada por ácido é a reação inversa da esterificação de Fischer.
A primeira etapa da reação é a ativação da carbonila por meio da protonação, ocorrendo o ataque nucleofílico pela água, formando o intermediário tetraédrico.
A protonação do –OR’ irá convertê-lo em um bom grupo abandonador e com isso ocorre a expulsão do álcool e a formação do ácido carboxílico.
Reações de ésteres
Como vimos no tópico anterior, os ésteres podem sofrer hidrólise catalisada por ácido, formando um ácido carboxílico, mas, também, sofrem hidrólise promovida por base, denominada saponificação (figura 9).

No refluxo de um éster em hidróxido de sódio aquoso, o íon hidróxido ataca a carbonila do éster para formar o intermediário tetraédrico. Então, a perda do íon alcóxido irá gerar um ácido carboxílico que será desprotonado pelo alcóxido formado, originando o íon carboxilato. O íon carboxilato, por ser pouco reativo, faz com que a hidrólise de um éster promovida por base seja uma reação essencialmente irreversível. Por fim, a protonação do íon carboxilato pela adição de um ácido em uma etapa separada forma o ácido carboxílico.
Já na presença de aminas primárias, secundárias ou amônia, os ésteres sofrem reação de substituição nucleofílica de acila, formando amidas. Entretanto, essa reação não é utilizada com frequência, pois é mais lenta do que as de cloretos de acila.
Os ésteres também podem ser sintetizados por meio de transesterificação, que nada mais é do que a reação em que se obtém um éster através de outro éster. O mecanismo dessa reação consiste simplesmente em adicionar um álcool e eliminar o outro álcool formado, ambas as etapas catalisadas por ácido.

No exemplo acima, podemos deslocar o equilíbrio para a direita, destilando da mistura o metanol – álcool de menor ponto de ebulição.
Saiba mais
O processo de transesterificação é importante para a geração de biodiesel a partir de óleos vegetais.
Reações de anidrido de ácido
Veja alguns exemplos dessas reações que resultam em medicamentos conhecidos:
Os anidridos de ácidos carboxílicos reagem com álcoois para formar ésteres, sem a necessidade de um catalisador ácido, como ocorre na esterificação (figura 11). O uso de um ácido forte pode ocasionar reações laterais, dependendo de quais outros grupos funcionais estejam presentes na molécula.
Um exemplo clássico desse tipo de reação é a síntese da aspirina (ácido acetilsalicílico) pela acetilação do
ácido o-hidroxibenzoico (ácido salicílico) com o anidrido acético.

Outra reação clássica utilizada pela indústria farmacêutica é conversão de anidrido acético em amida para a formação do Tylenol (acetominofeno). Nessa reação a p-hidroxianilina reage com o anidrido acético na presença base, em que o grupo -NH2, mais nucleofílico que o -OH, ataca a carboxila do anidrido.

A seguir, assista ao vídeo Reações de substituição nucleofílica acílica aplicadas à Indústria
Conversão de ácidos carboxílicos em amidas
As amidas podem ser sintetizadas utilizando-se vários métodos que envolvem a substituição nucleofílica de acila a partir de cloretos de ácidos, ésteres, anidridos (como visto nos itens anteriores), ácidos carboxílicos e sais carboxilatos.
A síntese de amidas a partir de ácidos carboxílicos é de difícil de preparo, pois as aminas utilizadas no método convertem os ácidos carboxílicos em seus ânions carboxilatos não reativos, não ocorrendo reação adicional em solução aquosa. Sendo assim, para que ocorra a formação de amidas, é necessária a evaporação da água e, subsequentemente, o aquecimento do sal seco, ocasionando a sua desidratação (figura 13).

Por ser um procedimento muito pobre, geralmente, o preparo de amidas é realizado a partir de cloretos de ácidos ou a partir de ácidos carboxílicos na presença de dicicloexilcarboiimida (DCC), que será responsável por ativá-lo (figura 14). A utilização de DCC na formação de amidas é a principal etapa de síntese laboratorial de pequenas proteínas ou peptídeos.

No mecanismo de reação mostrado na figura 15, a seguir, podemos observar que o ácido carboxílico irá se juntar à ligação C=N do DCC para que, posteriormente, ocorra a substituição nucleofílica de acila.

Reações de amidas
Assim como os cloretos de ácidos e ésteres, as amidas também sofrem hidrólise; entretanto, as condições necessárias são mais severas, normalmente necessitam de condições forçadas de aquecimento, e ácido ou base forte. A hidrólise ocorre quando as amidas em solução aquosa ácida ou básica são aquecidas, formando ácidos carboxílicos mais amônia ou amina. As etapas da hidrólise são reversíveis com o equilíbrio deslocado no sentido do produto.
Os mecanismos para a hidrólise ácida e básica estão descritos na figura 16. Na hidrólise ácida, a água age como um nucleófilo e ataca a amida protonada; posteriormente, ocorre a transferência de um próton do oxigênio ao nitrogênio, tornando-o um melhor grupo abandonador. Já a hidrólise básica é mais difícil de ocorrer, pois o íon amideto (-NH2) é um grupo abandonador fraco, dificultando a eliminação.

Verificando o aprendizado
ATENÇÃO!
Para desbloquear o próximo módulo, é necessário que você responda corretamente a uma das seguintes questões:
O conteúdo ainda não acabou.
Clique aqui e retorne para saber como desbloquear.
MÓDULO 3
Reconhecer as reações que envolvam a substituição do hidrogênio alfa (Hα) por eletrófilos e os princípios mecanísticos gerais da condensação de Claisen e da descarboxilação aplicada ao ácido malônico
Reações envolvendo a substituição do Hα por eletrófilos
Os derivados dos ácidos carboxílicos, principalmente em meio básico, podem ter o átomo de Hα substituído por eletrófilos, sendo intermediados pelos respectivos enolatos.
Antes de discutirmos melhor este assunto, vamos entender sobre a acidez dos hidrogênios α?
Ligações C-H em alcanos são pouco ácidas, pois o átomo de carbono não estabiliza o carbânion gerado, mas a presença de certos grupos funcionais, como ésteres e amidas, em posição adjacente à ligação C-H, irá influenciar muito no valor de pKa. Ou seja, quanto maior for a habilidade desses grupos em estabilizar a carga negativa da base conjugada por ressonância, maior será a acidez dos hidrogênios.
Na figura 1, podemos observar a forma de ressonância em que o oxigênio fica com a carga negativa – enolatos – é a que mais contribui para a dissociação da ligação C-Hα, pois o oxigênio é mais eletronegativo do que o carbono. Nesse caso, em que o hidrogênio α está entre duas carbonilas, o enolato será duplamente estabilizado, tornando-o mais estável.

Sendo assim, em ésteres e amidas terciárias, é possível gerar os respectivos enolatos por meio do tratamento com base, sendo que a posição do equilíbrio irá depender da força da base utilizada. A seguir, analisaremos as reações de condensação de Claisen e a síntese do éster malônico, que envolvem, justamente, a substituição do hidrogênio α.
Reação de condensação de Claisen
As reações de condensação de carbonila, de maneira geral, são muito encontradas nas rotas metabólicas para biossíntese de carboidratos, lipídeos, proteínas, ácidos nucleicos, entre outros, e têm grande importância, pois são um dos poucos métodos gerais para a formação de ligações carbono-carbono, possibilitando assim a construção de moléculas maiores a partir de precursores menores. (McMurry, 2011)
Dentre as reações de condensação, temos a reação de condensação de Claisen, que consiste no tratamento de um éster contendo pelo menos um átomo de hidrogênio na posição α ao grupo carboxílico com uma base, formando de maneira reversível o enolato correspondente. Na ausência de outras espécies eletrofílicas e em condições apropriadas, o enolato reagirá com o éster que lhe deu origem, formando o β-cetoéster correspondente. Ou seja, as condensações de Claisen envolvem o carbono α de uma molécula e o grupo carbonila de outra (figura 2).

Já a figura 3, que veremos a seguir, descreve o mecanismo da reação de condensação de Claisen, que envolve basicamente a adição nucleofílica de um íon enolato de éster no grupo carbonila de uma segunda molécula de éster.

No mecanismo de condensação, uma base alcóxido é utilizada para retirar um átomo de hidrogênio alfa (hidrogênio ácido) de uma molécula de éster, gerando um íon enolato nucleofílico.
A base alcóxido utilizada para formar o enolato deve ter o mesmo grupo alquila que o éster (etóxido para um éster de etila), pois, caso contrário, pode ocorrer a transesterificação que resultaria em mistura de cetoésteres ao final do processo.
O íon enolato, em uma reação de adição nucleofílica, irá atacar o carbono da carbonila de uma segunda molécula de éster, formando um intermediário alcóxido tetraédrico. O intermediário tetraédrico expulsará o íon etóxido, resultando na substituição do alcóxido pelo grupo derivado do enolato – formação do acetoacetato de etila.
Até aqui, se analisarmos em detalhes, veremos que o mecanismo de condensação de Claisen é um exemplo clássico de substituição nucleofílica de acila.
Entretanto, o equilíbrio global para o processo é desfavorável e pouco produto seria obtido se o processo envolvesse apenas essas duas etapas. Mas o íon etóxido é básico o suficiente para converter o produto β-cetoéster em seu respectivo ânion (base mais fraca) estabilizado por ressonância (etapa altamente favorável), deslocando assim o equilíbrio e completando a reação.
O álcool formado na reação, à medida que se forma, pode ser retirado por destilação, ajudando ainda mais o equilíbrio no sentido do produto desejado.
Por fim, ocorre a protonação do íon enolato pela adição de um ácido aquoso em uma etapa separada, formando o produto de condensação de Claisen.
Mas como sabemos que a desprotonação do β-cetoéster é a responsável por impulsionar a reação?
Se o éster utilizado no exemplo da figura 2 tivesse dois substituintes no carbono α (C2 do éster), o íon enolato estável não seria formado. Um éster com apenas um hidrogênio α não terá um hidrogênio ácido para que ocorra a terceira etapa do mecanismo e, como podemos esperar, o equilíbrio estará completamente desfavorável. Sendo assim, a reação não irá ocorrer sob as condições normais (EtO- em EtOH).
Entretanto, a reação pode ocorrer se uma base mais forte o suficiente para converter o éster em seu enolato – como, por exemplo, trifenilmetaneto de sódio – for utilizada. A reação do enolato com uma segunda molécula de éster formará o β-cetoéster com bom rendimento (figura 4). A segunda etapa da reação também poderia ser tratada com um cloreto de acila.

Condensações de Claisen intramoleculares
As condensações de Claisen intramoleculares, denominadas ciclização de Dieckmann, constituem-se em excelente método de síntese de cetonas cíclicas, particularmente ciclopentanonas e cicloexanonas. Na ciclização de Dieckmann, o átomo de carbono α e o grupo éster para condensação vêm da mesma molécula, em que um 1,6-diéster dá origem a um β-cetoéster cíclico com um anel de cinco membros, e a ciclização de um 1,7-diéster produz um β-cetoéster cíclico com um anel de seis membros. A formação de anéis menores é desfavorável devido à tensão angular do anel. Já os anéis maiores são entropicamente menos favoráveis devido ao maior número de conformações disponíveis para um precursor de cadeia mais longa. O mecanismo da ciclização de Dieckmann é semelhante ao da reação de Claisen (figura 5).

| Veja como funciona o mecanismo descrito na figura 5: |
|---|
| 1 - A base retira o próton ácido do carbono α de um dos dois grupos ésteres, gerando o íon enolato. |
| 2 - Em seguida, ocorre a adição nucleofílica intramolecular de um íon enolato ao grupo carbonila do éster da outra extremidade da cadeia, dando origem ao intermediário cíclico tetraédrico. |
| 3 - Ocorre a perda de um íon alcóxido do intermediário , isso leva à formação de um β-cetoéster cíclico, que será, posteriormente, desprotonado pelo alcóxido. |
| 4 - O íon enolato estável é então protonado pela adição de um ácido aquoso ao final da reação para gerar o β-cetoéster cíclico neutro. |
No exemplo a seguir (figura 6), podemos formar dois compostos β-cetoéster cíclico diferentes a partir de um mesmo éster. Na primeira reação, a 2-carboetoxiciclopentanona é sintetizada a partir do adipato de etila por refluxo em etanol na presença de etóxido de sódio e, por fim, o íon enolato é tratado com ácido aquoso. Já na segunda reação, o íon enolato formado é tratado com iodeto de etila, levando à formação da 2-etil-2-carboetoxiciclopentanona.

Condensações cruzadas de Claisen
A condensação cruzada de Claisen ocorre entre dois ésteres diferentes; entretanto, um deles não deve conter átomo de hidrogênio α e, consequentemente, será incapaz de formar um íon enolato e sofrer autocondensação.
Na figura 7, temos a reação do benzoato de etila com o acetato de etila. Nesse caso, o benzoato de etila não poderá produzir o íon enolato, comportando-se como receptor eletrofílico, ou seja, o parceiro de acilação.
Esse tipo de reação, porém, é limitado ao caso – além das restrições entre os tipos de ésteres – em que a adição do éster capaz de gerar o enolato é feita lentamente ao meio reacional contendo o éster não enolizável e a base. Dessa forma, será garantido um excesso do éster não enolizável durante a reação, evitando a autocondensação de Claisen do éster enolizável.

Para a realização desse tipo de condensação, existem vários tipos de ésteres reativos que não podem formar íons enolatos. Os quatro mais importantes estão descritos na figura 8.

Os três primeiros são mais eletrofílicos do que a maioria dos ésteres; sendo assim, são capazes de realizar a acilação em um enolato de forma mais rápida do que o éster que está sendo enolizado. Os oxalatos são muito reativos, pois cada grupo carbonila torna o outro mais eletrofílico. Nos formiatos, o átomo de hidrogênio os torna muito eletrofílicos, pois não há a conjugação σ e o impedimento estérico dos ésteres simples. Já os carbonatos são bastante úteis na introdução do grupo CO2R em um enolato. Por fim, os aromáticos serão os menos reativos por causa da conjugação do anel aromático, reduzindo a eletrofilicidade dos ésteres.
As reações de condensações de Claisen são acilações que sempre envolvem ésteres, mas enolatos de cetonas, por exemplo, podem funcionar tão bem quanto. Na reação a seguir (figura 9), entre um éster e uma cetona, apenas a cetona pode ser enolizada, e o éster utilizado, carbonato de dietila, é mais eletrofílico do que outra molécula de cetona.

No caso de cetonas assimétricas, um único produto será formado, pois a reação geralmente ocorre no lado menos substituído. No exemplo a seguir (figura 10), há a possibilidade de formação de dois produtos; entretanto, sob as condições de equilíbrio, apenas um deles pode formar um enolato estável.

Síntese do éster malônico
A síntese do éster malônico é um dos métodos mais antigos e úteis para alquilação de carbonilas. É um método utilizado para o preparo de ácidos carboxílicos empregando ésteres do ácido malônico (figura 11).

O éster malônico, também conhecido como malonato de dietila, contém hidrogênios α entre dois grupos carbonila, proporcionando maior acidez (pKa~13) frente aos compostos monocarbonílicos. Essa característica faz com que o éster malônico seja facilmente convertido em seu íon enolato mediante tratamento com bases moderadas, como alcóxidos de sódio ou potássio (pKa~16-18). O íon enolato, por ser um bom nucleófilo, reage rapidamente com haletos de alquila reativos (metila, primários, secundários, alílicos e benzílicos), gerando ésteres do ácido α-alquilmalônico. O mecanismo mais detalhado da reação é mostrado a seguir na figura 12.

Como mostrado na figura 12, o malonato de dietila forma um ânion enolato relativamente estável que pode ser alquilado em uma reação SN2. O produto dessa reação apresenta um átomo de hidrogênio α remanescente, de maneira que o processo de alquilação, sob condições básicas, pode ser repetido para formar um éster malônico dialquilado, se desejado, conforme mostra a figura 13.

A transformação de ésteres de ácidos malônicos mono- ou dissubstituídos nos ácidos monocarboxílicos correspondentes ocorre por meio da hidrólise ácida ou básica para os respectivos derivados de ácido malônico, e os ácidos malônicos substituídos descarboxilam-se rapidamente.

A descarboxilação não é uma reação geral de ácidos carboxílicos; pelo ao contrário, ela só pode ocorrer em compostos que apresentam um segundo grupo carbonila na posição β do ácido. Isto é, apenas os ácidos malônicos e os β-cetoácidos sofrem perda de –CO2 sob aquecimento. Usando esse método, os β-cetoácidos fornecem cetonas e os ácidos malônicos fornecem ácidos carboxílico simples (ocorre apenas uma descarboxilização). Observe na figura 15, a seguir, que a descarboxilização ocorre por um mecanismo cíclico e envolve a formação inicial de um enol.

No vídeo que você verá agora, poderá se aprofundar na Síntese do éster malônico: síntese de ácidos acéticos substituídos.
Síntese de éster malônico utilizando dialoalcanos
Neste item iremos mostrar dois exemplos da síntese de éster malônico que fazem uso de dialoalcanos.
Na síntese do ácido glutárico, dois equivalentes em quantidade de matéria do sal de sódio do éster malônico reagem com um dialoalcano. Nesse caso, ocorrerão duas alquilações consecutivas, originando um tetraéster. Em seguida, ocorrerá a hidrólise e a descarboxilação do tetraéster, fornecendo um ácido dicarboxílico.

No segundo exemplo, veremos que a síntese do éster malônico também pode ser usada na preparação dos ácidos cicloalcanocarboxílicos, a partir de um dialoalcano. Veja a seguir como isso acontece:
Utilizar um equivalente em quantidade de matéria do sal do éster malônico para reagir com um equivalente de um dialoalcano, fornecendo um éster haloalquilmalônico.
Etapa de alquilação: ocorre de maneira intramolecular, gerando um produto cíclico.
Ocorre a hidrólise e a decarboxilação para fornecer o produto, ácido ciclopentanocarboxílico.
Esse método é frequentemente utilizado para preparar anéis de três, quatro, cinco e seis membros; entretanto, o rendimento diminui para anéis maiores.

Biossíntese de ácidos graxos
Como mencionamos no início do módulo, as reações de condensação de carbonila, de maneira geral, são muito encontradas nas rotas metabólicas, como, por exemplo, a formação de ácidos graxos utilizando a condensação de Claisen para a formação de ligações C-C. A maioria dos ácidos graxos naturais é constituída por um número par de átomos de carbono, reforçando a ideia, apresentada em 1893 e confirmada muitos anos depois, de que sejam montados a partir de unidades de acetato (CH3CO2-). (SOLOMONS & FRYHLE, 2006)
Hoje, sabe-se que a biossíntese de ácidos graxos começa com a acetil-coenzima A, um tioéster da coenzima A (figura 18).

Antes de a síntese de ácidos graxos começar, porém, devemos observar a transferência dos grupos acila da acetilCoA e MalonilCoA para outros grupos tiois presentes no sistema enzimático responsável pela biossíntese de ácidos graxos (figura 19).

Em seguida, a ausência de espécies básicas no meio fisiológico é suprida pela descarboxilação do maloniltioéster (figura 20), garantindo a formação do enlato necessário para realização da condensação de Claisen. A condensação do acetil-SR e do malonil-SR é altamente exotérmica, e a posição de equilíbrio localiza-se bem para a direita, sendo termodinamicamente favorável porque a reação produz o dióxido de carbono, substância altamente estável. Por fim, com o enolato formado, espécie nucleofílica, ocorre a adição à carboxila do tioéster derivado da acetilCoA, como explicado por COSTA, 2005.

Após a formação do β-cetoéster, ocorre uma série de reações (reduções e desidratação) para formação de um novo éster com quatro átomos de carbono na sua cadeia principal (figura 21). Esse novo éster sofre uma série de reações semelhantes às descritas anteriormente, agora formando um novo tioéster contendo seis átomos de carbono na cadeia principal.

Essa sequência de reações é repetida até que o tioéster apresente uma quantidade apropriada de carbono, lembrando que a cada repetição há um aumento de dois carbonos na cadeia principal. Por fim, esse tioéster sofrerá transesterificação com o glicerol para formar óleos, gorduras e fosfolipídeos essenciais ao metabolismo.
Neste módulo vimos que, assim como a condensação de Claisen, a descarboxilção representa um papel importante nas rotas metabólicas, principalmente na síntese de ácidos graxos.
Verificando o aprendizado
ATENÇÃO!
Para desbloquear o próximo módulo, é necessário que você responda corretamente a uma das seguintes questões:
O conteúdo ainda não acabou.
Clique aqui e retorne para saber como desbloquear.
Conclusão
Considerações Finais
Neste tema, abordamos as principais reações envolvendo ácidos carboxílicos e seus derivados, e discutimos os princípios mecanísticos de cada uma, destacando sua importância no entendimento da Química Orgânica.
Por fim, apresentamos as principais fontes e rotas metabólicas desses compostos. Ao concluir o estudo deste tema, você deve ter percebido que os ácidos carboxílicos e seus derivados apresentam rotas e usos bastantes diversificados e participam da rotina dos laboratórios e indústrias.